有機半導体の電荷移動のしやすさの評価: Wannier解析を用いたトランスファー積分の算出#

有機半導体や導電性高分子において、マクロな「電荷移動度(モビリティ)」はデバイス性能を決定づける最重要パラメータです。その微視的な支配要因となるのが、隣接する分子間で電荷がどれくらい移動しやすいかを示す「トランスファー積分(Transfer Integral)」です。本事例では、第一原理計算ソフトウェアAdvance/PHASEによる高精度な電子状態計算と、Wannier90を連携させた最大局在ワニエ関数(MLWF)の構築により、代表的な有機半導体であるペンタセン(Pentacene)結晶のトランスファー積分を算出し、その顕著な伝導異方性を評価した事例を紹介します。
Keywords: 第一原理計算 (DFT), Wannier90, ペンタセン (Pentacene), トランスファー積分 (移動積分), 電荷移動度, 異方性
理論的背景と導出#
トランスファー積分とマーカス理論#
高温領域(室温など)における半古典的なホッピング伝導モデル(Marcus Theory)において、サイト間の電荷移動速度定数 は、再配向エネルギー と、分子間の電子的相互作用であるトランスファー積分 を用いて次のように表されます [1]。
ここで、 はプランク定数、 はボルツマン定数、 は温度です。移動速度定数はトランスファー積分の2乗に比例します()。電荷の移動しやすさを理解し、デバイス設計の指針を得るためには、 の定量的かつ高精度な評価が不可欠です。
周期境界条件(PBC)を満たす結晶系において、波数空間で広がったブロッホ関数をユニタリ変換し、実空間の各分子サイトに局在化したワニエ関数(Wannier Function)[2] を構築することで、タイトバインディング(強束縛)モデルにおけるハミルトニアンの非対角要素として を一義的に抽出することができます。
p型有機半導体のホール伝導とHOMO軌道#
ペンタセンなどの有機半導体は、電界効果トランジスタ(OFET)などで広く利用されています。高純度なペンタセン結晶においては、電子もホール(正孔)と同等に高い移動度を持つことが示されていますが [3]、実際のOFETデバイスの多くはp型(ホール伝導)として動作させます。そのため、デバイス設計の観点からは、ホール伝導の経路となる隣接分子間の最高被占軌道(HOMO)の重なりを正確に評価することが重要となります。
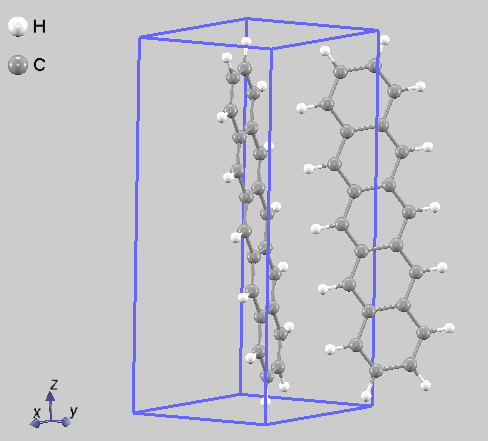
図1. ペンタセン結晶の単位胞構造モデル。特徴的なヘリンボーン配列が確認できます。
計算方法と技術的な工夫#
本解析は、ペンタセン結晶(図1)を対象とします。計算コストの抑制と精度の両立を目指し、以下のワークフローで実施しました。
- 剛体シミュレーションによる構造探索の高速化:
分子性結晶において、弱い分子間力によってポテンシャルエネルギー曲面が平坦かつ複雑になることに加え、分子の回転など膨大な自由度があります。既存事例 [4] では、最初にペンタセン分子を剛体として扱ったシミュレーションを行いました。この剛体最適化で得られた良好な初期構造から全原子の緩和計算を行うことで、安定構造を効率よく決定しました。 - 高密度k点メッシュによる非自己無撞着(Non-SCF)計算:
SCF計算でペンタセン結晶の電子密度分布を決定した後に、滑らかな波動関数を得るために10x8x4の均等で高密度なk点メッシュを用いてNon-SCF計算を実施しました。 - Wannier化のための初期射影(重心)の決定:
Wannier90でHOMO軌道を各分子に正しく局在化させるためには、初期射影の中心を分子の「重心」に置くことが重要です。周期境界条件(セル境界)をまたぐ分子を正しく処理するため、Pythonスクリプトを用いて分子の重心位置を自動算出しました。 - HOMOバンドのピンポイント抽出(
exclude_bandsの活用):
ペンタセン結晶の単位胞には2つの分子が含まれており、HOMO由来のバンドは2本存在します。多数の価電子バンドの中から、このホール伝導の主役である「HOMO由来の2本のバンド」のみをターゲットとするため、Wannier90の入力に除外設定(exclude_bands)を組み込みました。これにより、計算コストを抑えて高速なインターフェース処理 [5] を実現しています。
計算結果と考察#
抽出されたトランスファー積分と伝導の異方性#
Wannier90の反復計算は良好に収束し、実空間の強結合ハミルトニアン(_hr.dat)を出力しました。出力データから、基準となる分子から周囲の分子へのホッピングエネルギー()の絶対値を抽出した結果を図2および表1に示します。
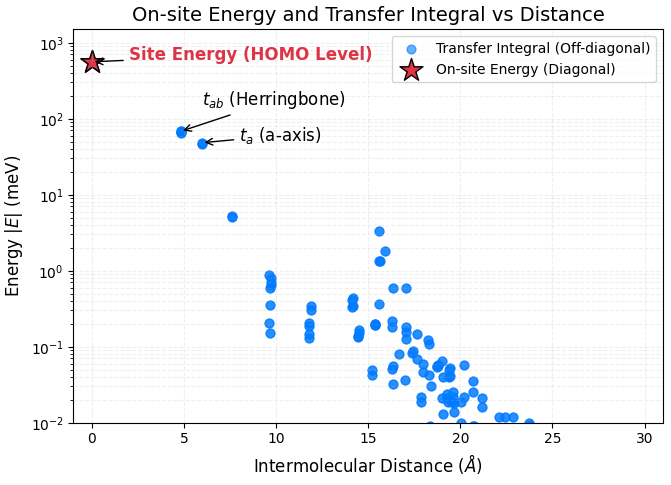
図2. ペンタセン結晶におけるエネルギー成分(オンサイトエネルギーおよびトランスファー積分の絶対値)と分子間距離の関係。距離ゼロ付近の赤い星印は分子自身のHOMO準位(オンサイトエネルギー)を表し、青い丸印は他分子への電荷移動のしやすさを示すトランスファー積分を表しています。主要な伝導パス(\(t_{ab}\) および \(t_a\))に強い相互作用が集中し、距離が離れるにつれて指数関数的に減衰する様子が確認できます。
表1. ワニエ解析により得られたペンタセン結晶のトランスファー積分
| 移動方向(パス) | 絶対値 (meV) |
|---|---|
| 斜め方向 () | 約 65.5 ~ 68.2 |
| a軸方向 () | 約 48.3 |
| b軸方向 () | 約 5.2 |
| c軸方向 () | 約 0.37 |
既存文献との比較とデバイス設計への示唆#
得られた数値から、ペンタセン結晶における電荷移動のダイナミクスについて以下の重要な知見が得られます。
- 特徴的な2次元伝導系: 層間方向(c軸方向)のトランスファー積分 が 0.4 meV 未満と極端に小さいのに対し、面内方向(, )は 50 meV 近い大きな値を示しました。これは、ホールがa-b面内(2次元層内)に強く閉じ込められて移動することを裏付けています。
- 既存文献との一致: Cornilら [3] によるINDO法(半経験的分子軌道法)を用いた先行研究では、a軸方向のトランスファー積分は meV と評価されています。また、Coropceanuら [6] の報告でも斜め方向の相互作用が最大であることが示されています。今回の第一原理計算とWannier化に基づく算出結果( meV, meV)は、これらの文献値と非常に良い一致を示しており、本ワークフローの信頼性が裏付けられました。
これらの結果は、有機FETを作製する際に「電極をどの結晶軸に対して配置すれば最も高い移動度が得られるか」というデバイス設計の指針を提供します。
まとめ#
本事例では、第一原理計算ソフトウェアAdvance/PHASEと外部の解析パッケージWannier90の連携機能を駆使することで、有機結晶系におけるトランスファー積分の計算ワークフローを実証しました。ペンタセン結晶を対象として、バンドの絞り込み(除外設定)や重心の定義といった技術的アプローチにより、計算コストを抑えつつ既存の文献値とよく一致するトランスファー積分を算出しました。
本解析の詳細や、研究への適用可能性に関するご相談はこちら
お問い合わせ参考文献#
- J. L. Brédas, J. P. Calbert, D. A. da Silva Filho, and J. Cornil, "Organic semiconductors: A theoretical characterization of the basic parameters governing charge transport", PNAS 99, 5804 (2002).
- N. Marzari, A. A. Mostofi, J. R. Yates, I. Souza, and D. Vanderbilt, "Maximally localized Wannier functions: Theory and applications", Rev. Mod. Phys. 84, 1419 (2012).
- J. Cornil, J. Ph. Calbert, and J. L. Brédas, "Electronic Structure of the Pentacene Single Crystal: Relation to Transport Properties", J. Am. Chem. Soc. 123, 1250 (2001).
- 剛体シミュレーションの活用:分子性結晶の構造探索の高速化
- Wannier局在軌道の解析・Wannier90連携による化学結合と電子状態の解釈
- V. Coropceanu, J. Cornil, D. A. da Silva Filho, Y. Olivier, R. Silbey, and J.-L. Brédas, "Charge Transport in Organic Semiconductors", Chem. Rev. 107, 926 (2007).
関連ページ#
- 第一原理計算ソフトウェア Advance/PHASE
- 解析分野:ナノ・バイオ
- 産業分野:材料・化学