剛体シミュレーションの活用:分子性結晶の構造探索の高速化#

分子性結晶は、次世代の材料として大きな注目を集めています。特に、有機半導体、有機伝導体、そして全固体電池といった先端技術分野で、その有用性が大いに期待されています。第一原理計算ソフトウェアAdvance/PHASEには、分子性結晶の構造探索に特に有用である剛体シミュレーションの方法が実装されています。ここでは、その方法の解析事例を紹介します。
Keywords: 第一原理計算 (DFT)、分子性結晶、全固体電池、高速化、剛体シミュレーション
分子性結晶について#
分子性結晶(クラスター結晶を含む)は、分子内では原子が強く結合している一方、分子間の相互作用は弱いという特徴があります [1]。第一原理計算において、この弱い分子間力を標準的な手法で正確に記述することは難しく、分子性結晶の物性予測や構造決定が困難であることはよく知られています。そのため、第一原理シミュレーションで分子性結晶の構造探索を効率的に行うには、計算手法に特別な工夫が求められます。
Advance/PHASEは、指定した原子グループを一つの剛体 [2] とみなし、その重心に働く並進力とトルクに基づいて構造緩和や分子動力学(MD)シミュレーションを実行する「剛体束縛機能」を備えています。この機能を用いることで、通常の計算手法では時間がかかりがちな分子性結晶の構造緩和や、表面への分子吸着といった系の計算を大幅に高速化することが可能です。
解析モデルと解析結果#
Advance/PHASEのGUIでは、分子性結晶に対して各原子の分子属性の自動判定が可能です。この機能を用いて各原子がどの分子に属するかを自動的に判別します。そして、原子の「剛体への割り当て」を行い、剛体シミュレーションを実行できます。
図1は、分子性結晶である白燐の構造最適化を実施した例を示しています。このような時間スケールの異なる、分子内と分子間モードが混在する系の場合、計算の途中で剛体ダイナミクスを活用すること (CG法 → 剛体ダイナミクス → CG法) によって総計算時間を大幅に短縮することがわかります。また、最後の収束にはCG法を使用することで完全な構造緩和(full relaxation)を達成しました。
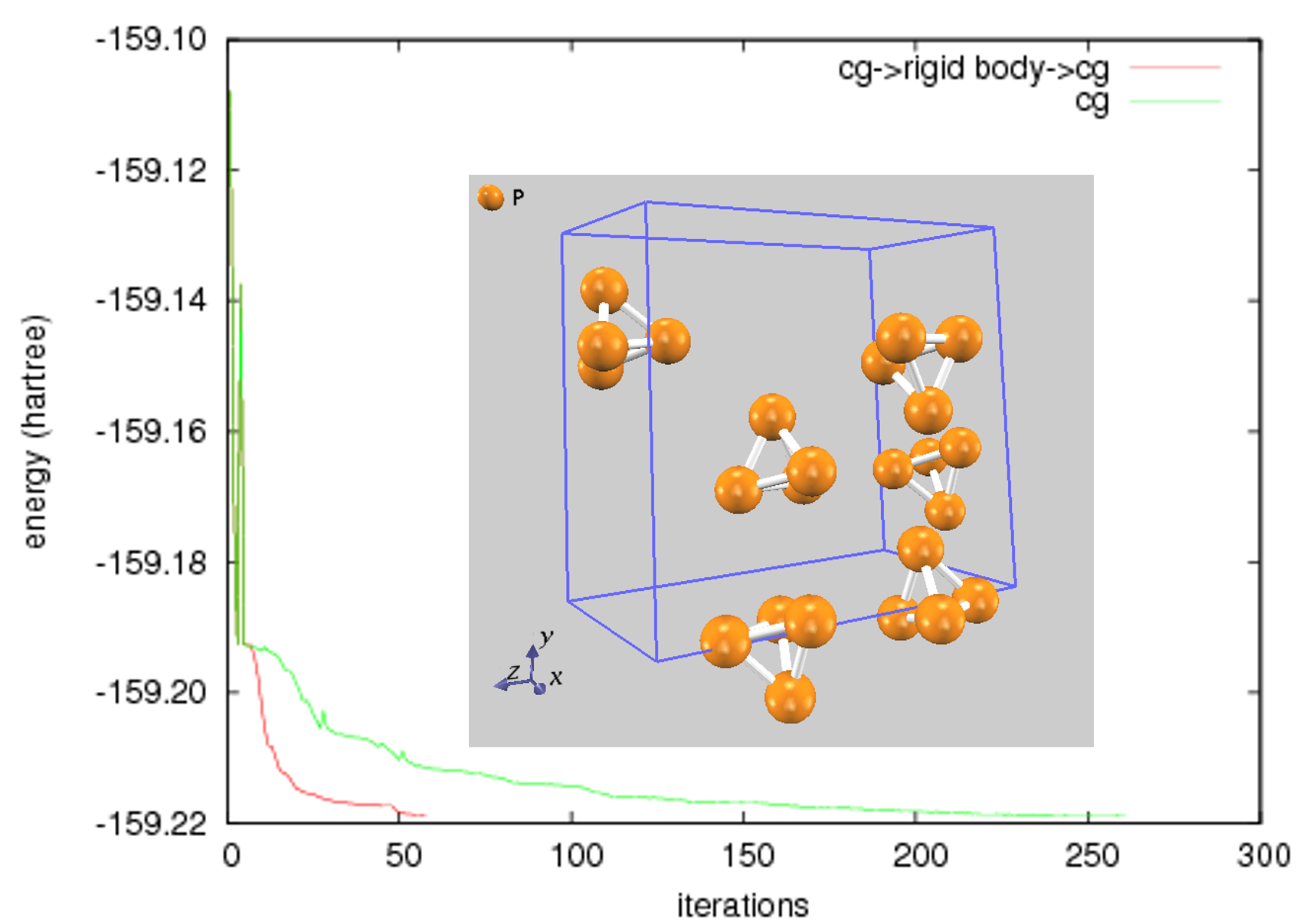
図1.白燐の構造最適化におけるエネルギーの履歴。赤線:途中で剛体ダイナミクスを利用した場合,緑線:最初から最後までCG法を利用した場合 (Inset: 計算における最初の原子配置)
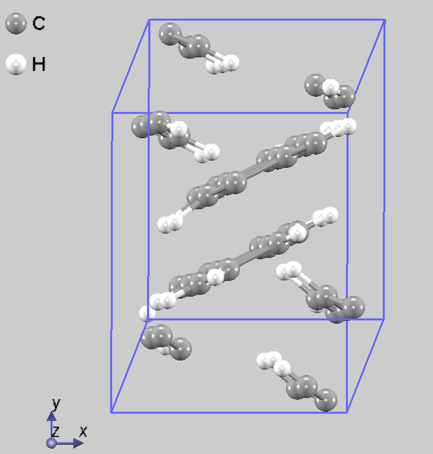
図2にペンタセン結晶の結果を示します。計算の開始からfull relaxationで構造最適化を行うと24並列で1日計算しても収束する様子が見られませんでした。一方、最初にペンタセン分子を剛体として扱ったシミュレーションを行うと206回の繰り返し計算で収束し、かかった時間は24並列で1時間弱でした。その後剛体シミュレーションからfull relaxationに切り替えると45回の繰り返し計算で収束し、かかった時間は24並列で20分弱となりました。結果として2時間以内に構造最適化できました。
(a) モデル構造
(b) 剛体シミュレーションでの変化
(c) Full relaxationでの変化
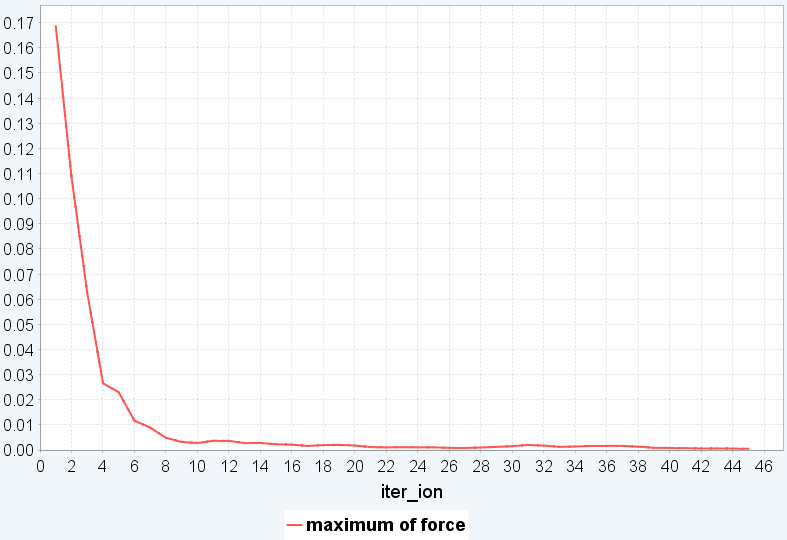
図2.ペンタセン結晶の構造最適化 (a)モデル構造(b)剛体シミュレーションの繰り返し計算での最大力の変化(c)剛体からfull relaxationに切り替えた後の繰り返し計算での最大力の変化
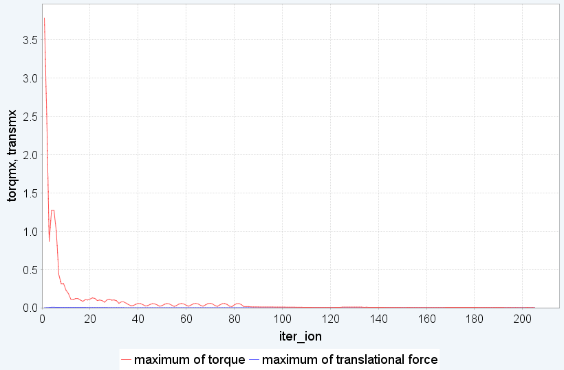
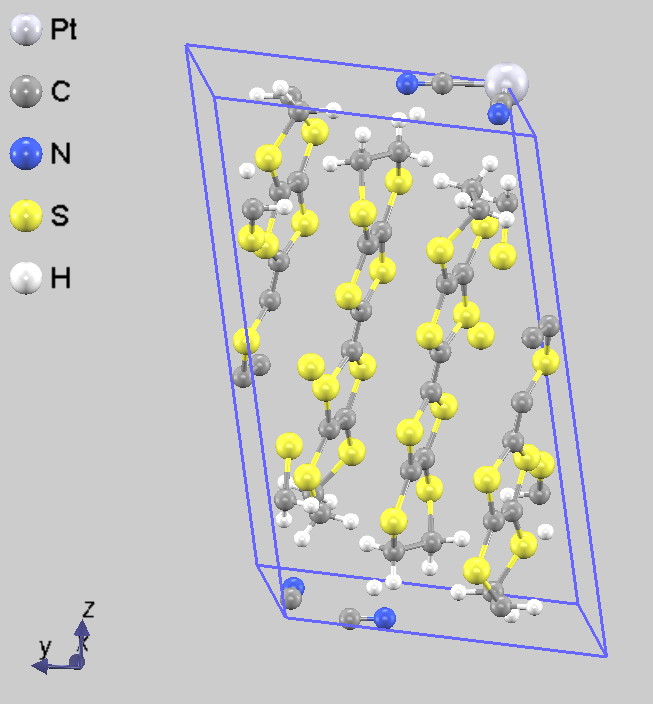
図3は BEDT-TTF_PtNO の構造最適化の結果を示しています [BEDT-TTF = Bis(ethylenedithio)tetrathiafulvalene ビス(エチレンジチオ)テトラチアフルバレン]。まず、それぞれの分子を剛体として扱ったシミュレーションを行うと52回の繰り返し計算で収束し、かかった時間は24並列で1.5時間程度でした。その後剛体シミュレーションからfull relaxationに切り替えると97回の繰り返し計算で収束し、かかった時間は12並列で3時間弱となりました。
(a) モデル構造
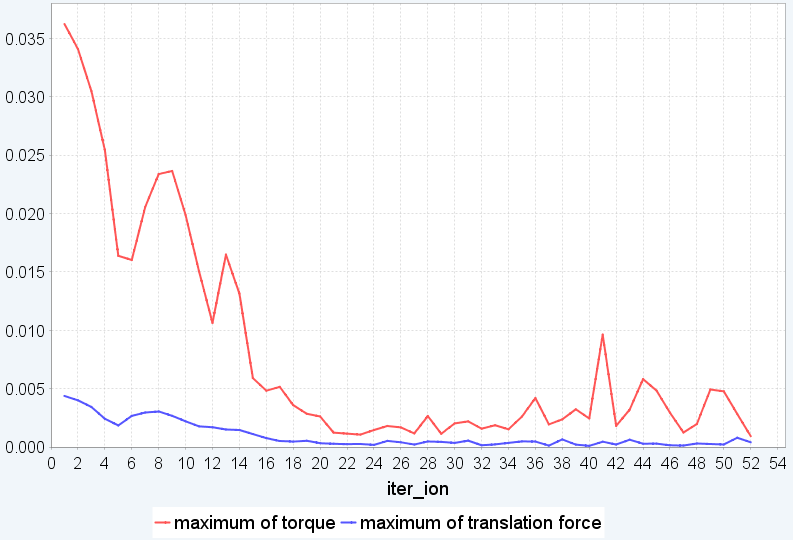
(b) 剛体シミュレーションでの変化
(c) Full relaxationでの変化
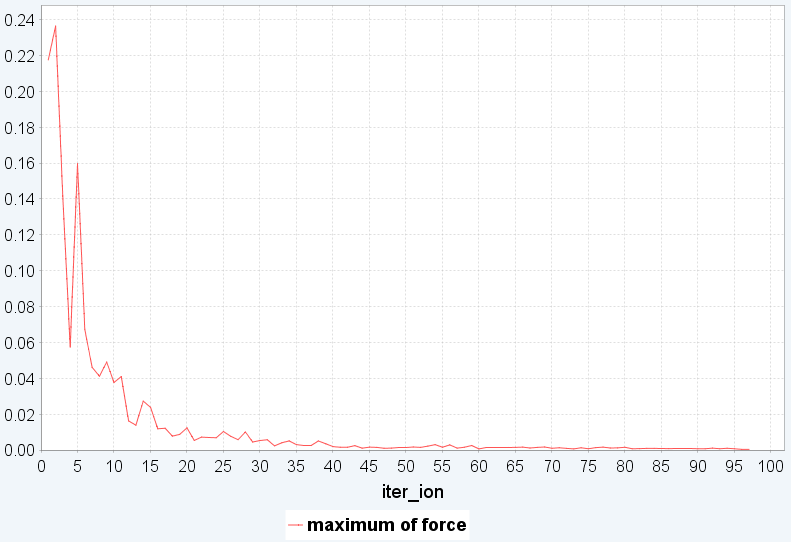
図3. BEDT-TTF_PtNO の構造最適化。(a)モデル構造、(b)剛体シミュレーションの繰り返し計算でのmaximum torque, maximum translational forceの変化、(c)剛体からfull relaxationに切り替えた後の繰り返し計算での最大力の変化
有機伝導体への応用事例として、(BEDT-TTF)2I3の計算結果を紹介します。安定構造を求めた後にバンド計算を行い、フェルミ面より上の部分領域に対応する電荷密度を求めると、図4で示すように、結晶の縦軸方向に電荷密度がつながり、横軸方向にはつながっていない電荷密度が得られました。これは、縦軸方向は伝導的、横軸方向は非伝導的であることを示しており、実際の物性を正しく再現しています。
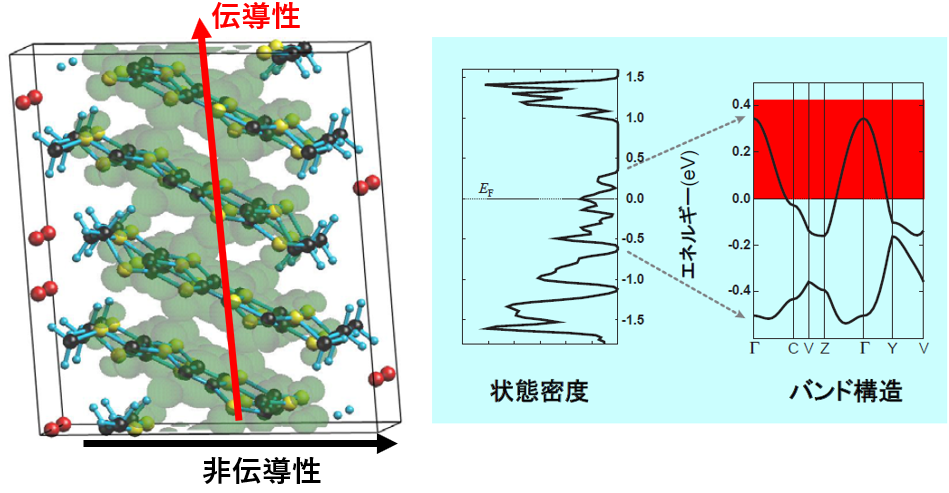
図4.(BEDT-TTF)2I3の電荷密度分布、状態密度、およびバンド構造
まとめ#
Advance/PHASEに実装された剛体シミュレーション機能は、様々な分子性結晶の構造最適化を大幅に高速化します。分子性結晶の構造最適化は、通常、困難を伴います。これは、弱い分子間力によってポテンシャルエネルギー曲面が平坦かつ複雑になることに加え、分子の回転など膨大な自由度を持つためです。この問題を解決する有効な手法が二段階法です。まず、分子を一つの剛体とみなし、最適な配置と向きを効率的に探索します。次に、この剛体最適化で得られた良好な初期構造から全原子の緩和計算を行うことで、計算が安定し、真の安定構造へと効率的に収束させることが可能になります。これは、複雑な問題を「粗視的 → 詳細的」なアプローチで解く、非常に強力な戦略です。
本解析の詳細や、研究への適用可能性に関するご相談はこちら
お問い合わせ参考文献#
- Kitaigorodsky, A. L. Molecular crystals and molecules. Vol. 29. Elsevier, 2012.
- G. Ciccotti and J.P. Ryckaert, Computer Physics Reports 4 (1986) 346.
関連ページ#
- 第一原理計算ソフトウェア Advance/PHASE
- 解析分野:ナノ・バイオ
- 産業分野:材料・化学