AI(LightGBM)と第一原理計算を用いた誘電率(電子・格子)の予測と検証#

半導体・絶縁体材料の設計において、全誘電率は素子の性能を左右する基幹的な物性です。全誘電率は、電場に対する電子雲の応答である「電子分極」に起因する電子誘電率(高周波誘電率 )と、イオンの変位による応答である「格子分極」に起因する格子誘電率(イオン誘電率 )の和として表され、両者は物理的な起源が異なります。本事例では、近年の「AI for Science」を具現化する取り組みとして、機械学習を用いて高速に誘電率を予測し(仮想スクリーニング)、その結果を第一原理計算で裏付ける「マテリアルズ・インフォマティクス(MI)」の二段構えのアプローチを実践しました。勾配ブースティング決定木「LightGBM」を主軸としたスタッキングモデルを用いて、Materials Project の誘電率データベースから電子誘電率・格子誘電率をそれぞれ独立に予測し、抽出した代表的な5物質(α-SiO2, AlN, GaN, ZnO, LiTaO3)について第一原理計算ソフトウェア Advance/PHASE による誘電率の検証を行いました。電子誘電率と格子誘電率で予測の難易度が大きく異なること、そしてその差がなぜ生じるのかを、AIが捉えた物理的特徴とともに考察します。
Keywords: 第一原理計算 (DFT), マテリアルズ・インフォマティクス (MI), LightGBM, スタッキング, 誘電率, 電子分極, 格子分極
1. 機械学習モデルの構築と予測#
LightGBMとスタッキングについて#
LightGBM(Light Gradient Boosting Machine)は、複数の決定木(Decision Tree)を直列に組み合わせて強力な予測モデルを構築する「勾配ブースティング」アルゴリズムを、ヒストグラムベースの高速化手法によって極限まで効率化した機械学習ライブラリです [1]。マテリアルズ・インフォマティクスで主流となる表形式(テーブル)の材料データに対しては、深層学習と同等以上の高い予測精度と安定性を、はるかに少ない計算コストで発揮します。
本事例では予測精度をさらに高めるため、性質の異なる複数のベースモデル(LightGBM・Extra Trees・Ridge回帰)の予測を、上位のメタ学習器で統合する「スタッキング(Stacking)」[2] を採用しました。メタ学習器には、過学習を防ぎやすいRidge回帰(交差検証によるハイパーパラメータ最適化付き)を用いています。決定木系と線形系では誤差の傾向が異なるため、これらを組み合わせることで単一モデルの弱点を補い合い、汎化性能の向上が期待できます。
誘電率の分解と予測戦略
全誘電率は電子誘電率と格子誘電率の和()で表されます。両者はデータの分布が大きく異なり、電子誘電率は真空の誘電率を下限としておおむね2〜10の範囲に収まる一方、格子誘電率は0からフォノンの軟化によって数十以上の巨大な値まで広く分布します。そこで本事例では、電子誘電率と格子誘電率を独立した2つのターゲットとして別々に学習し、桁の広い物性値を扱うため対数空間(log1p変換)で回帰する設計としました。
データの準備と特徴量エンジニアリング#
LightGBMの学習データとして、Materials Project(MP)[3] から誘電率(電子・格子)およびバンドギャップが既知である無機化合物データを取得しました。金属(バンドギャップがゼロの物質)を除外し、さらに計算が破綻した外れ値や強誘電体近傍の極端な巨大値を除く前処理(電子誘電率 1.0 ≤ ε∞ < 30、格子誘電率 0 ≤ εion < 100)を施した結果、約6,800件のデータセットを構築しました。これを学習用(85%)とテスト用(15%)に分割し、汎化性能を評価する設計としています。最終的なターゲットとなる5つの候補物質は、真の予測能力を検証するため学習・テストデータ群から完全に除外(ブラインドテスト化)しました。
取得した結晶構造と化学組成に対し、オープンソースライブラリ matminer [4] を用いて、元素ごとの原子体積・価電子数・電気陰性度などの統計的特徴量(Magpie特徴量)と、密度・充填率・空間群などの構造記述子を自動生成しました。さらに、誘電率の物理に直結する以下の特徴量を独自に追加しています。これらは予測精度の向上に大きく寄与しました。
- 電子誘電率予測向け:バンドギャップ とその逆数 、および (ペンモデル に基づく。電子誘電率はバンドギャップと強く相関する)
- 格子誘電率予測向け:極性空間群フラグ・反転対称性の有無(強誘電性=巨大な格子誘電率の代理指標)、電気陰性度の広がり(ボルン有効電荷の代理)、平均原子質量(フォノン振動数の代理)
ハイパーパラメータ最適化と予測精度#
ハイパーパラメータ最適化ライブラリ Optuna [5] を用いて、電子誘電率・格子誘電率それぞれに最適なパラメータを独立に探索しました。テストデータに対する予測精度は、電子誘電率で 決定係数 (R2 Score): 0.92(MAE: 0.51)、格子誘電率で R2 Score: 0.44(MAE: 3.6)となりました。電子誘電率については、化学組成や構造から得られるスカラー記述子ベースの予測モデルでありながら、高い精度(実用レベル)を達成しています。一方、格子誘電率の精度が電子誘電率に大きく劣る点は、後述するように物理的な要因によるものであり、本事例の重要な論点となります。
表1. 機械学習モデルの予測精度(テストデータ評価)
| 予測対象 | 決定係数 R2 | 平均絶対誤差 MAE | 予測の難易度 |
|---|---|---|---|
| 電子誘電率 ε∞ | 0.92 | 0.51 | 比較的容易 |
| 格子誘電率 εion | 0.44 | 3.6 | 困難 |
特徴量の重要度と物理的解釈#
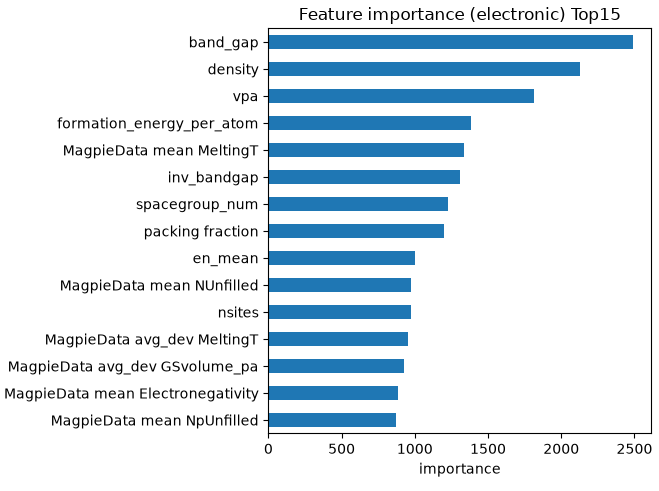
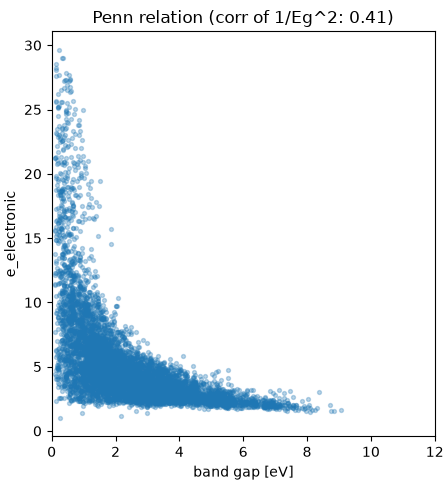
図1. (上)電子誘電率予測における特徴量重要度 Top15。(下)バンドギャップと電子誘電率の関係(ペンモデルの確認)。
図1(上)は、電子誘電率の予測においてモデルが根拠とした特徴量の重要度を示しています。バンドギャップ(band_gap)が最も重要な特徴量として選ばれており、その逆数(inv_bandgap)や平均電気陰性度(en_mean)も上位に入っています。これは、電子誘電率がバンドギャップと密接に関係するという物理(ペンモデル)を、AIがデータから正しく捉えていることを意味します。
図1(下)のバンドギャップと電子誘電率の散布図を見ると、バンドギャップが小さい物質ほど電子誘電率が大きくなるという反比例の傾向(ペンモデルの予言)が明確に現れています。ただし同じバンドギャップでも電子誘電率には相当なばらつきがあり( との相関係数はおおむね0.41)、バンドギャップ単独では決まりきらない部分を、密度・原子体積・構造記述子といった他の特徴量が補完することで、高い予測精度が実現されています。
一方、格子誘電率の予測では、極性・非極性を区別する空間群番号(spacegroup_num)が最重要特徴量となり、バンドギャップがそれに次ぎました。格子分極はイオンの変位(フォノン)に由来するため、結晶の対称性(極性の有無)が決定的に重要であるという物理が、ここにも反映されています。
2. 第一原理計算での検証#
次に、機械学習が算出した予測値の「答え合わせ」を行うため、Advance/PHASE を用いた第一原理計算 (DFT)の結果 [6] と比較しました。Advance/PHASE では、自己無撞着場(SCF)計算で得られた電子状態から電子誘電率(高周波誘電率 )を、格子振動解析とベリー位相法によるボルン有効電荷(Z*)の計算から格子誘電率()をそれぞれ直接求めることができます。なお誘電率は本来テンソル量であり、DFT計算でも c 軸に垂直な方向(xx)と平行な方向(zz)で異なる値が得られますが、本稿では機械学習によるスカラー予測値との比較のため、方向平均した等方値 を用いています。
表2. 各手法による誘電率の比較(電子・格子・全体、いずれも方向平均値)
| 物質 | 電子誘電率 ε∞ | 格子誘電率 εion | 全誘電率 εtotal | 実験値 (静誘電率) |
|||
|---|---|---|---|---|---|---|---|
| ML予測 | DFT | ML予測 | DFT | ML予測 | DFT | ||
| α-SiO2 | 2.6 | 2.3 | 1.8 | 2.4 | 4.4 | 4.7 | ~4.5 |
| AlN | 4.7 | 4.5 | 4.0 | 4.1 | 8.7 | 8.6 | ~9 |
| GaN | 6.3 | 5.3 | 4.7 | 4.4 | 11.0 | 9.7 | ~9.8 |
| ZnO | 6.3 | 4.2 | 4.6 | 5.7 | 10.9 | 9.9 | ~8.2 |
| LiTaO3 | 5.4 | 4.4 | 19.8 | 40.2 | 25.2 | 44.6 | ~42 |
※ DFT計算値は c 軸垂直(xx)・平行(zz)成分を方向平均した等方値。ML予測はスカラー値。実験値は方向平均の概数。
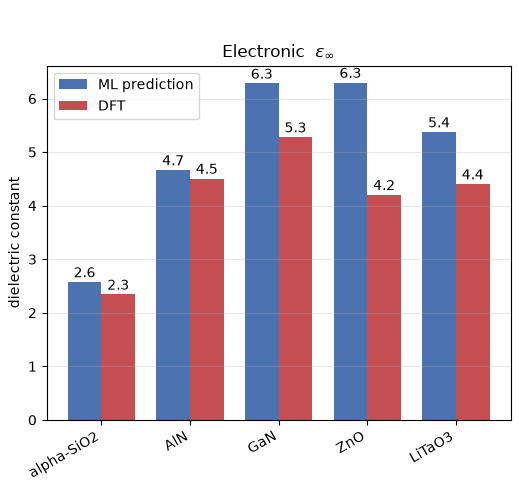
図2. 機械学習による電子誘電率の予測値とDFT計算による参照値の比較。
予測と検証の考察#
図2(電子誘電率)および表2(全成分)の比較から、いくつかの重要な傾向が読み取れます。
第一に、電子誘電率についてはML予測とDFT計算値が良好な一致を示しており、特に α-SiO2 や AlN では優れた一致を示しています。全体としてMLの予測値がわずかに大きめに出る傾向がありますが、これはMP教師データに含まれる計算(GGA-PBE 系)がバンドギャップを過小評価しがちな性質を反映したものと考えられます。なかでも ZnO においては差異が大きく、これは d 電子を持つ酸化物に特有の難しさによるものです。興味深いことに、Advance/PHASE による直接計算でも ZnO の電子誘電率は実験値に対して過大評価する傾向が報告されており、これも同じく GGA-PBE がバンドギャップを過小評価することに起因します。機械学習と第一原理計算が、同じ物理的要因から同じ方向のずれを示している点は示唆に富みます。
第二に、格子誘電率では、強誘電体である LiTaO3 においてMLが大幅な過小評価を示しました。Advance/PHASE の計算によれば、LiTaO3 の巨大な格子誘電率は、Ta 原子が公称価数(+5)を大きく超える +7 以上の異常に大きなボルン有効電荷を持ち、これが特定の格子振動モード(ソフトモード)と結合することに由来します [6]。しかし機械学習は「組成と平衡構造の平均的な特徴量」から学習しているため、このようなフォノンの軟化に起因する特異な巨大応答を捉えることができず、予測値が中央(平均寄り)に引っ張られます。
本解析における格子誘電率の予測精度(R2 Score: 0.44)を踏まえると、今後は組成や全体構造から計算されたスカラー特徴量に頼るだけでなく、結晶構造そのものの局所的なトポロジーや原子間相互作用を直接学習できるGraph Neural Network (GNN) の導入が望ましいと考えられます。
まとめ#
本事例では、データベースとLightGBM(スタッキング)を用いた「高速な誘電率予測」と、Advance/PHASEによる「高精度な第一原理検証」を組み合わせたハイブリッドな物性予測アプローチを実践しました。電子誘電率(高周波誘電率)は、バンドギャップをはじめとする物理に基づく特徴量を活用することで、高精度(R2 = 0.92、実用レベル)で予測できることを示しました。一方、格子誘電率は、フォノンの軟化が支配する強誘電体においては、本事例に用いたモデルでの予測が困難であり、まさにこうした物質においてこそ第一原理計算による検証が不可欠であることを明らかにしました。第一原理計算は、単に誘電率の値を高精度に再現するだけでなく、ボルン有効電荷の解析を通じて、LiTaO3 の巨大な格子誘電率が Ta 原子の異常ボルン有効電荷(+7超)とソフトモードの結合に由来するという物理的起源まで解明できます。AI(機械学習)が広大な探索空間から候補を高速に絞り込み、第一原理計算がその物性のミクロな起源を明らかにする——データ科学と計算科学の「両輪」を活用し、それぞれの得意領域を組み合わせることで、材料開発のコストと時間を大幅に削減することが可能です。
本解析の詳細や、研究への適用可能性に関するご相談はこちら
お問い合わせ参考文献#
- G. Ke, Q. Meng, T. Finley, T. Wang, W. Chen, W. Ma, Q. Ye, T.-Y. Liu, "LightGBM: A Highly Efficient Gradient Boosting Decision Tree", Advances in Neural Information Processing Systems 30, 3146 (2017).
- D. H. Wolpert, "Stacked Generalization", Neural Networks 5, 241 (1992).
- A. Jain et al., "Commentary: The Materials Project: A materials genome approach to accelerating materials innovation", APL Materials 1, 011002 (2013).
- L. Ward et al., "Matminer: An open source toolkit for materials data mining", Comput. Mater. Sci. 152, 60 (2018).
- T. Akiba, S. Sano, T. Yanase, T. Ohta, and M. Koyama, "Optuna: A Next-generation Hyperparameter Optimization Framework", Proceedings of the 25th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, 2623 (2019).
- 誘電関数の第一原理計算:各種材料の解析事例
関連ページ#
- 第一原理計算ソフトウェア Advance/PHASE
- 解析分野:ナノ・バイオ
- 産業分野:材料・化学