GCMC法との連携:Mg-MOF-74の高精度CO2吸着シミュレーションと分極効果#

多孔性配位高分子(MOF)のガス吸着性能を理論的に予測・評価する上で、ミクロな電子状態の知見をマクロな熱力学特性へとスケールアップするマルチスケール解析が注目されています。本解析では、第一原理計算ソフトウェアAdvance/PHASEにより算出された高精度な最適化構造と部分電荷を力場(Force Field)として活用し、グランドカノニカル・モンテカルロ(GCMC)法によるCO2吸着等温線の予測シミュレーションへと連携させる事例を紹介します。
Keywords: 第一原理計算 (DFT), GCMC, RASPA, Mg-MOF-74, CO2吸着, DDEC電荷, 分極力場 (Polarizable Force Field)
DDEC部分電荷を活用したGCMC法の理論的背景#
GCMC法と静電相互作用の重要性#
グランドカノニカル・モンテカルロ(GCMC)法 [1] は、一定の温度()、体積()、および化学ポテンシャル()の下で、粒子(吸着質分子)の挿入、削除、並進、回転を確率的に行うことで、実験的な吸着平衡状態を統計力学的にシミュレートする手法です。各モンテカルロステップにおける状態遷移の受理確率 (acceptance probability) は、メトロポリス法に基づき以下のように決定されます(粒子の挿入・削除の例)。
粒子の挿入 (Insertion):
$$P_\text{acc} = \min \left[ 1, \frac{V}{\Lambda^3 (N+1)} \exp(\beta \mu) \exp(-\beta \Delta U) \right]$$
粒子の削除 (Deletion):
$$P_\text{acc} = \min \left[ 1, \frac{\Lambda^3 N}{V} \exp(-\beta \mu) \exp(-\beta \Delta U) \right]$$
ここで、\(\Lambda\) は熱的ド・ブロイ波長、\(N\) は粒子数、\(\beta = 1/k_BT\)、\(\Delta U\) は遷移に伴う系のポテンシャルエネルギー変化です。
実際のシミュレーションでは、化学ポテンシャル は系の圧力(より厳密にはフガシティー )と の関係で結びついています。これにより、任意の実験圧力に対応するマクロな吸着量をコンピュータ上で再現することが可能となります。
そして、ポテンシャルエネルギー変化 は、ファンデルワールス相互作用(通常はLennard-Jonesポテンシャル)と静電相互作用、および分極エネルギーの和()として計算されます。
MOFのような複雑な多孔性材料において、ゲスト分子(特に四重極モーメントを持つCO2など)と骨格間の静電相互作用を正確に評価することは極めて重要です。静電相互作用エネルギー はクーロンの法則により計算されます。
第一原理計算から算出されるDDEC(Density Derived Electrostatic and Chemical)電荷 [2] をこの部分電荷 として用いることで、空間の静電ポテンシャル(ESP)を物理的に妥当な形で高精度に再現できます。
オープンメタルサイトにおける分極効果の重要性#
Mg-MOF-74は1次元の六角形細孔を持ち、その壁面に配位不飽和な金属サイト(Open Metal Site: OMS)であるMg2+が規則的に配列しています。CO2のような四重極モーメントを持つ分子がこのOMSに接近すると、Mg2+の強い局所電場によってCO2の電子雲が歪み、誘起双極子(分極)が生じます。
本事例で適用した分極力場では、この分極による誘導エネルギー を明示的に計算します。分子内の各サイト に誘起される双極子 と、周囲の電荷(DDEC電荷を含む)が作る静電場 との相互作用は次のように表されます。
ここで、\(\alpha_i\) は原子の分極率です。バックポラリゼーション(誘起双極子同士の相互作用)を無視することで、計算コストを抑えつつ効率的にGCMC法に組み込むことが可能になります。
計算手法:DFTとGCMCのマルチスケール連携#
本解析では、多孔性材料の吸着シミュレーションに特化したオープンソースコード RASPA2 [3] を用いました。シミュレーションの構築は以下のステップで行いました。
- 構造と電荷のインポート: Advance/PHASEで得られた最適化座標と各原子のDDEC電荷(Mgサイト: 約+1.41 e)[4] を剛体骨格として定義します。
- 力場の割り当て: MOF骨格のファンデルワールス力にはUFF(Universal Force Field)を、CO2分子には気液平衡を正確に再現するTraPPEモデルを採用します。
- 分極(Polarization)の導入: OMSの強力な電場による相互作用を正確に評価するため、Beckerら [5] が提案した「分極力場(Polarizable FF)」を適用しました。LJパラメータ()を意図的にスケールダウンして暗黙の分極効果を排除し、代わりに各原子に固有の分極率()を割り当てています。
- 静電場と誘起双極子の自己無撞着な計算: RASPA2の機能(
ComputePolarization yes)を用い、各モンテカルロステップにおいて周囲の電場に応じた誘起双極子を計算し、静電相互作用を直接的に評価しました。なお、計算コストの観点から高次のバックポラリゼーションは無視する設定としています。
計算結果と考察#
1. 標準力場(UFF)の限界と分極効果(Polarization)#
温度298 K、圧力10 kPa(0.1 bar)というCO2回収プロセスにおいて実用上重要な低圧条件にて、GCMCシミュレーションを実施しました。
当初、標準的なUFFパラメータとDDEC電荷のみを用いた計算(固定電荷モデル)では、絶対吸着量が約0.91 mol/kgと、実験値(約5.3~6.3 mol/kg)[4] を大幅に過小評価しました。これは、剛体モデルのファンデルワールス反発力が強すぎ、オープンメタルサイトの静電引力が十分に発揮される前にCO2が弾かれてしまうためです。
そこで、金属サイト特有の強い相互作用を正確に記述するため、分極(Polarization)効果を明示的に組み込んだ計算を実施しました。結果は以下の通りです。
表1. GCMCシミュレーションによるCO2吸着特性の比較(298 K, 10 kPa)
| 評価モデル | 絶対吸着量 (mol/kg) | 吸着熱 (-, kJ/mol) |
|---|---|---|
| 標準UFF + DDEC電荷 | 0.91 ± 0.06 | 26.0 ± 0.5 |
| 分極力場(Polarizable FF) + DDEC電荷 | 6.42 ± 0.18 | 39.4 ± 2.2 |
| 実験値の目安 | 約 5.3 ~ 6.3 | 約 40.6 ~ 43.2 |
分極効果を導入した結果、吸着熱が39.4 kJ/molに上昇し、吸着量は6.42 mol/kgへと大幅に改善しました。エネルギー解析の結果、分極による引力エネルギーは純粋なクーロン引力に匹敵する大きな寄与を示しており、極性を持たない分子(CO2)であっても、局所的な強電場下では電子雲の分極が吸着の主要な駆動力になることが定量的に確認されました。
2. 吸着サイトの可視化#
シミュレーションで得られた吸着平衡状態のスナップショットを図1に示します。CO2分子が細孔内を無秩序に分布するのではなく、壁面、特に六角形の頂点(Mgサイト)に強固に吸着(局在化)している様子が分かります。これはMgのオープンメタルサイトが正しく強力な引力を発揮している証拠です。CO2は「酸素(O) - 炭素(C) - 酸素(O)」という直線分子ですが、CO2の端にある酸素原子が、MOFのMg原子に向かって直接配向して配置されています。酸素のマイナス電荷とMgのプラス電荷が引き合っているこの「End-on配向」は、分極力場とDDEC電荷を正しく設定したからこそ再現できた現実の挙動そのものです。
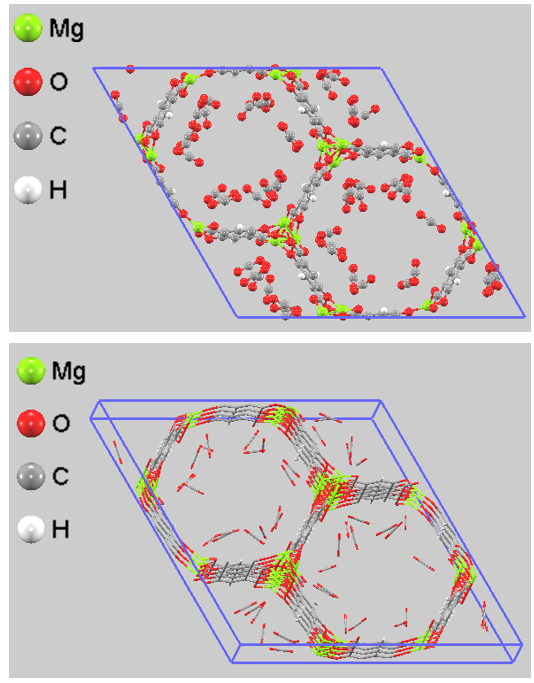
図1. 分極力場を用いたGCMCシミュレーションによるMg-MOF-74のCO2吸着スナップショット(上:正面図、ball-and-stick; 下:斜視図、bond only)。六角形細孔の壁面に露出したMgサイトに対して、分極によって強力な引力を受けたCO2分子が吸着している様子が確認できます。
まとめ#
本事例では、第一原理計算ソフトウェアAdvance/PHASEによるDFT計算(ミクロな電子状態・DDEC電荷)と、GCMC法(マクロな熱力学・統計力学)を連携させて、CO2吸着等温線の予測シミュレーション(温度298 K、圧力10 kPa)を実施しました。特に、オープンメタルサイトのような特異な環境においては、標準力場をそのまま適用するのではなく、物理的洞察に基づき「分極効果」などの適切な相互作用モデルを選択することが、精度の高い物性予測において非常に重要であることが示されました。
本解析の詳細や、研究への適用可能性に関するご相談はこちら
お問い合わせ参考文献#
- D. Frenkel and B. Smit, "Understanding Molecular Simulation: From Algorithms to Applications", 2nd ed., Academic Press (2001).
- T. A. Manz and N. G. Limas, "Introducing DDEC6 atomic population analysis: part 1. Charge partitioning theory and methodology," RSC Adv. 6, 47771 (2016).
- D. Dubbeldam, S. Calero, D. E. Ellis, and R. Q. Snurr, "RASPA: molecular simulation software for adsorption and diffusion in flexible nanoporous materials," Mol. Simul. 42, 81 (2016).
- 多孔性配位高分子Mg-MOF-74の構造・細孔解析およびDDEC部分電荷の評価
- T. M. Becker, J. Heinen, D. Dubbeldam, L.-C. Lin, and T. J. H. Vlugt, "Polarizable Force Fields for CO2 and CH4 Adsorption in M-MOF-74," J. Phys. Chem. C 121, 4659 (2017).
関連ページ#
- 第一原理計算ソフトウェア Advance/PHASE
- 解析分野:ナノ・バイオ
- 産業分野:材料・化学