多孔性配位高分子Mg-MOF-74の構造・細孔解析およびDDEC部分電荷の評価#

多孔性配位高分子(MOF: Metal-Organic Framework)は、金属イオンと有機配位子がネットワークを形成し、内部にナノスケールの空間を有する材料です。 中でもMg-MOF-74(Mg/DOBDC、別名: CPO-27-Mg)は、細孔表面に露出した「不飽和金属サイト(オープンメタルサイト)」を持ち、二酸化炭素(CO2)などの小分子に対して驚異的な吸着性能を示すことで知られています。 本事例では、第一原理計算ソフトウェアAdvance/PHASEを用いてMg-MOF-74の最適化構造を決定し、幾何学的な細孔解析と、DDEC法による高精度な部分電荷・結合次数の評価を連携して行った結果をご紹介します。これらの評価は、MOFの吸着性能を判断する重要な指標となります。
Keywords: 第一原理計算 (DFT), Mg-MOF-74, オープンメタルサイト, CO2吸着, DDEC電荷, 結合次数, Zeo++, 分散力補正
計算手法:多角的なアプローチによるMOFの評価#
MOFの吸着性能は、「分子が入るための空間の広さ(幾何学的特性)」と「分子を引き寄せる静電気力(電子的特性)」の両方に支配されます。本解析では、以下の3ステップで多角的な評価を実施しました。
- 構造最適化: Advance/PHASEを用い、分散力相互作用を考慮したPBE+DFT-D3(BJ)汎関数にてセル構造および内部座標の最適化を行いました。MOFのような多孔質材料では、骨格を支える分散力の記述が不可欠です。
- 細孔解析: 最適化された構造に対し、外部ツールZeo++ [1] を用いて、細孔径、比表面積、細孔体積などの幾何学的な評価を行いました。
- DDEC電荷と結合次数の解析: 第一原理計算から価電子密度のcubeファイルを出力し、外部プログラムChargemol [2] と連携させることで、静電ポテンシャルを高精度に再現するDDEC6部分電荷および結合次数 (Bond Order) を算出しました。
計算結果と考察#
1. 構造最適化と粉末X線回折(XRD)シミュレーション#
PBE+DFT-D3(BJ)を用いて最適化されたMg-MOF-74の結晶構造をもとに、粉末XRDパターンのシミュレーションを行いました。 図1に示すように、低角側(2θ = 6.8°付近および11.7°付近)にMOF-74特有の強い回折ピーク [3] が確認されました。
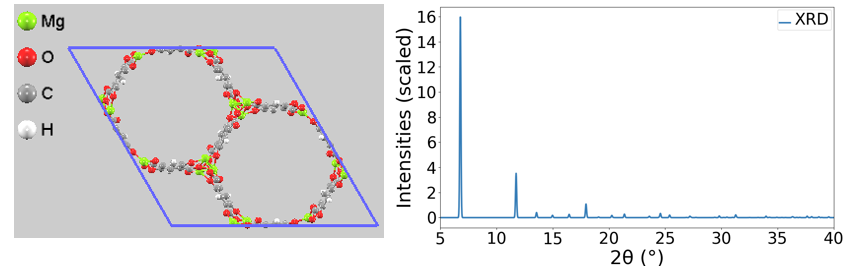
図1. 最適化構造(左)から計算されたMg-MOF-74の粉末XRDシミュレーションパターン(右)
2. Zeo++による幾何学的・細孔特性の評価#
空間的な広さがCO2の吸着・拡散に十分であるかを確認するため、Zeo++により細孔解析を実施しました。プローブ半径としてN2相当(1.82 Å)および、純粋な空洞サイズを測るための 0.0 Å を使用しました。
表1. Mg-MOF-74の幾何学的特性の計算結果
| 評価項目 | 計算値 | 物性的意味 |
|---|---|---|
| 最大空洞直径 (LCD) | 11.82 Å | 細孔内部で最も広い空間のサイズ |
| 細孔限界直径 (PLD) | 11.07 Å | 分子が拡散する際の「ボトルネック」のサイズ |
| 質量あたり表面積 (ASA) | 1687.7 m2/g | N2プローブがアクセス可能な表面積 |
| 質量あたり細孔体積 (N2プローブ) |
0.359 cm3/g | プローブ分子の中心がアクセス可能な体積 |
| 質量あたり幾何学的体積 (プローブ半径 0 Å) |
0.748 cm3/g | 骨格原子が占有しない純粋な空洞の総体積 (上限値) |
LCDとPLDがほぼ等しい値(約11〜12 Å)を示しており、一直線に貫通した1次元トンネル構造であることが数値的にも裏付けられました。また、質量あたり表面積(ASA)は 1687.7 m2/g と算出され、実験的に報告されているLangmuir比表面積(1741 m2/g [3])と非常に良い一致を示しました。
一方で、質量あたりの細孔体積については、N2の剛体球モデルを仮定したアクセス可能体積が 0.359 cm3/g と算出されました。実験的に報告されている細孔体積(0.573 cm3/g [3])と比較すると、やや小さめに見積もられています。これは、第一原理計算による完全結晶の剛体モデル(原子の熱振動や骨格の柔軟性を考慮しない静的な構造)を用いていることや、実際の合成サンプルに存在しうる構造欠陥(ミッシングリンカー等)が計算モデルには含まれていないことに起因すると推測されます。とはいえ、空洞の幾何学的な総体積(プローブ半径 0 Å)は 0.748 cm3/g と十分な空間を確保しており、LCDや表面積の良好な一致も踏まえれば、ガス吸着メカニズムを解析するための構造モデルとしては妥当であると言えます。
3. DDEC部分電荷による活性サイトの可視化#
次に、CO2を引き寄せる「駆動力」を評価するため、Chargemolを用いてDDEC(Density Derived Electrostatic and Chemical)法による部分電荷解析を実施しました。
MOF/COFシミュレーションにおけるDDEC部分電荷解析の優位性
第一原理計算から部分電荷を求める手法にはMulliken法やBader法、RESP法など様々な種類がありますが、多孔性材料(MOF/COF)の解析においてはDDEC法が優れた適性を示します。その理由は以下の通りです。
- 静電ポテンシャル(ESP)の正確な再現: MOFのガス吸着(特にCO2やH2Oなどの極性分子)は、骨格とゲスト分子間の静電相互作用に強く支配されます。 DDEC電荷は、空間の静電ポテンシャルを物理的に妥当な形で再現するように最適化されるため、GCMC(グランドカノニカル・モンテカルロ)法などを用いた高精度なガス吸着シミュレーション用の力場(Force Field)構築に最適です。
- 「埋もれた原子」に対する物理的妥当性: 表面の静電ポテンシャルのみにフィッティングする手法(RESP法など)は、MOFのような複雑な3Dネットワークの内部(表面に露出していない原子)の電荷が物理的に無意味な値に発散しやすい弱点があります。DDEC法は電子密度に立脚しているため、すべての原子に対して化学的直感(酸化数や電気陰性度)に合致する安定した電荷を与えます。
- 周期境界条件(PBC)への対応: 分子モデルだけでなく、MOFのような無限に続く結晶の周期境界系に対してもシームレスかつ高精度に適用可能です。
このDDEC法を用いて計算されたMg-MOF-74の各原子の部分電荷は以下の通りです。
表2. 各原子のDDEC部分電荷
| サイト | 元素 | DDEC電荷 (e) |
|---|---|---|
| Mg1 | Mg | +1.41 |
| O1, O2, O3 | O | -0.79 〜 -0.64 |
| C1, C2 | C | -0.20 〜 +0.68 |
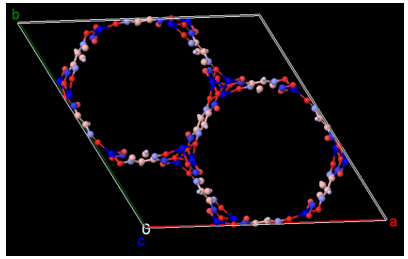
図2. DDEC電荷の可視化(Jmol [4] 使用。正電荷:青、負電荷:赤)
表2および図2から明らかなように、細孔の壁面に露出したMg原子は +1.41 e という強い正電荷を帯びています。この値が形式電荷(+2.0)より値が小さいことは、酸素配位子との間に適度な電子共有(共有結合性)が存在して骨格を安定化させていることを示しています。同時に、このむき出しの正電荷(ルイス酸点)こそが、四重極モーメントを持つCO2分子の酸素原子(負電荷)を強力に引き寄せる「磁石」の正体です。
4. 結合次数 (Bond Order) 解析による配位状態の解明#
最適化された結晶構造から、Mgサイトは5つの酸素原子と配位結合した不飽和状態(5配位)であり、細孔内に「オープンメタルサイト」を形成していることが確認できます。この特異な配位環境において、DDEC解析によって算出されたMgの結合次数の総和(SBO: Sum of Bond Orders)は約 1.33 を示しました。
一般に、MgとOの結合はイオン性が強いとされますが、個別のMg-O間の結合次数をみると平均 0.26 程度の値を持っており、完全なイオン結合ではなく、電子軌道の重なりによる適度な共有結合性が混在していることが分かります。この「5配位による構造的な不飽和性」と「適度な共有結合性による骨格の安定化」が両立していることこそが、Mg-MOF-74が高いCO2吸着性能を発揮する電子的・構造的な基盤となっています。
まとめ#
本解析では、第一原理計算ソフトウェアAdvance/PHASEを用いて、DFT計算と外部ツールの連携が、MOFの物性解明において有効であることが示されました。幾何学的解析 (Zeo++) と 電子的解析 (DDEC6)を組み合わせることで、空間的余裕と静電的引力の両面から吸着メカニズムを定量的・視覚的に説明することが可能になりました。この解析ワークフローは、Mg-MOF-74にとどまらず、新たなガス分離材、触媒、蓄電池材料など、様々な先端材料の「活性サイトの特定」や「吸着メカニズムの解明」に応用可能です。
本解析の詳細や、研究への適用可能性に関するご相談はこちら
お問い合わせ参考文献#
- T. F. Willems, C. H. Rycroft, M. Kazi, J. C. Meza, and M. Haranczyk, "Algorithms and tools for high-throughput geometry-based analysis of crystalline porous materials," Microporous and Mesoporous Materials 149, 134 (2012).
- T. A. Manz and N. G. Limas, "Introducing DDEC6 atomic population analysis: part 1. Charge partitioning theory and methodology," RSC Adv. 6, 47771 (2016).
- S. R. Caskey, A. G. Wong-Foy, and A. J. Matzger, "Dramatic Tuning of Carbon Dioxide Uptake via Metal Substitution in a Coordination Polymer with Cylindrical Pores," J. Am. Chem. Soc. 130, 10870 (2008).
- http://jmol.sourceforge.net/
関連ページ#
- 第一原理計算ソフトウェア Advance/PHASE
- 解析分野:ナノ・バイオ
- 産業分野:材料・化学