Mo(110)表面へのN2分子の吸着:DFT計算を用いた基礎検討#

アンモニア(NH3)合成は、現代の化学工業において最も重要なプロセスの一つであり、その効率化には、触媒表面で窒素分子(N2)の強固な三重結合がいかに解離するかが鍵となります。本解析では、第一原理計算ソフトウェアAdvance/PHASEを用いた密度汎関数理論(DFT)計算によって、代表的なアンモニア合成触媒の活性成分であるモリブデン(Mo)の最密充填面(110)を対象に、N2分子の吸着挙動を調査しました。本検討は「基礎検討」として、将来的な反応障壁計算(NEB法)やより精密な触媒活性予測に向けた前段階と位置付けられており、分子状吸着と解離吸着のどちらがエネルギー的に有利であるかを物理的・化学的観点から明らかにすることを目的としています。
Keywords: 第一原理計算, DFTシミュレーション, 表面吸着, アンモニア合成触媒, モリブデン, 窒素分子, 分子状吸着, 解離吸着, スラブモデル
計算モデルと計算条件#
Mo(体心立方構造)の(110)面を再現するため、3x3の直交スーパーセル(長方形スーパーセル) を用いた5層のスラブモデルを構築しました。表面現象を正確に記述するため、スラブの底面2層を固定し、残りの層および吸着分子を自由に動かして構造最適化を行っています。真空層の厚さは17 Å以上としました。交換相関汎関数には標準的なPBE(GGA)を採用し、信頼性のある基礎検討を目指しました。
表1. 計算条件の概要
| 項目 | 設定内容 |
|---|---|
| 計算手法 | 平面波・擬ポテンシャル ※ウルトラソフト型擬ポテンシャル使用 |
| 交換相関汎関数 | GGA (PBE) |
| カットオフエネルギー | 25 Ry |
| モデル設定 | Mo(110) 3x3 直交スーパーセル, 5層モデル(底面2層固定) |
| 真空層厚 | 十分な厚さを確保 (> 17 Å) |
| k点サンプリング | 3x2x1 |
| 構造最適化閾値 | 1x10-3 Hartree/bohr |
計算結果:吸着構造とエネルギー#
4つの吸着配置の比較#
本解析では、N2分子の吸着サイトや方位が異なる4つの代表的な初期配置(Config 1~4)を選定しました。図1に、それぞれの構造最適化後の原子配列を示します。なお、固定層の原子は半透明で表示しています。
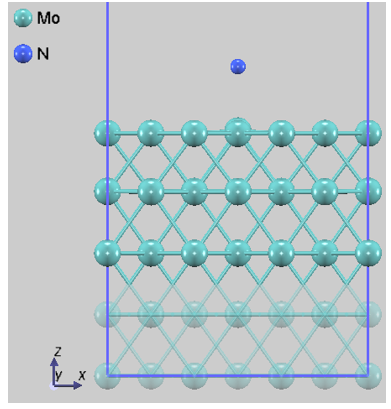
図1a. Config 1
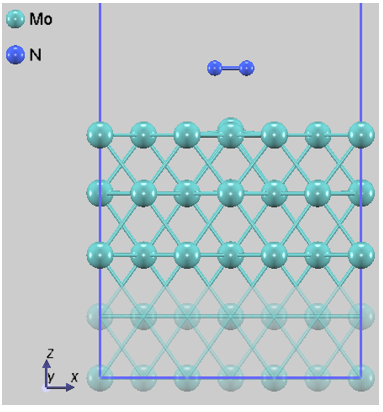
図1b. Config 2
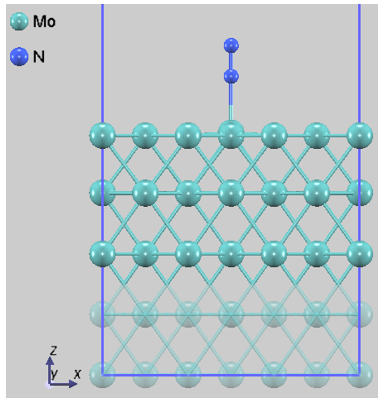
図1c. Config 3 (分子状吸着)
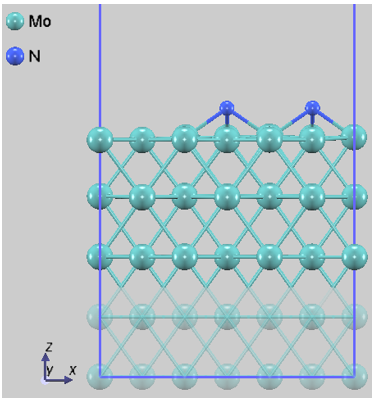
図1d. Config 4 (解離吸着)
図1. Mo(110)表面におけるN2の各吸着配置。Config 3は分子が直立して表面に結合しており、Config 4ではN-N結合が切断され、原子が表面のくぼみに配置されています。
各配置における吸着エネルギー()は、以下の式を用いて算出しました。
ここで、 はN2分子が吸着した系全体の全エネルギー、 は清浄なMo(110)スラブの全エネルギー、 は孤立したN2分子の全エネルギーを表します。本定義により、値が負に大きくなるほど、より安定な吸着であることを示します。計算結果と構造パラメータを表2にまとめます。
表2. 吸着特性の算出結果
| Config | 吸着エネルギー Ead [eV] | 最近接Mo-N距離 [Å] | N-N距離 [Å] | 状態の判別 |
|---|---|---|---|---|
| 1 | 0.03 | 2.41 | 1.16 | エネルギー的に不安定 |
| 2 | 0.04 | 2.36 | 1.16 | エネルギー的に不安定 |
| 3 | -0.67 | 2.09 | 1.14 | 準安定(分子状吸着) |
| 4 | -3.50 | 1.97 | 5.54 | 最安定(解離吸着) |
※PBE汎関数を用いているため分散力が考慮されず、物理吸着の安定化エネルギーが得られていません。
考察:分子吸着から解離吸着へ#
計算結果より、Mo(110)表面上では窒素分子が原子状に解離した「解離吸着状態(Config 4)」が、エネルギー的に最安定(Ead = -3.50 eV)となることが明らかになりました。この吸着エネルギーは、分子状吸着状態と比較して約2.8 eVも低く、表面上では解離状態が熱力学的に強く支配的であることを示しており、先行研究 [1] とも整合しています。
一方、分子状吸着(Config 3)におけるN-N結合長は1.14 Åと算出され、気相中の結合長(1.117 Å)に対し伸長が見られることから、本構造は解離反応の前駆体(precursor state)として機能していることが示唆されます。解離吸着によって得られる大きなエネルギー利得は、Mo表面がN≡N三重結合の切断に対して高い活性を有することを理論的に裏付けるものです。ただし、過度に強力な吸着相互作用は、生成物であるアンモニアの脱離過程を律速させる要因ともなり得るため、実用触媒としての評価においては吸着・脱離のエネルギーバランス(Sabatierの原理)への考慮が不可欠です。
なお、本計算における解離吸着(Config 4)は、解離した2つのN原子が近接して存在する「共吸着状態」です。原子が表面拡散して互いに離れた「孤立吸着状態(単原子吸着×2)」と比較すると、原子間の横方向相互作用(主に反発力)の分だけエネルギーが高くなる可能性がありますが、反応直後の生成系としては本モデルが妥当と考えられます。
更なる検討と触媒活性予測への展望#
本検討により基礎的な吸着傾向が判明しましたが、実用的な触媒開発に向けては以下の発展的なアプローチが必要です。
1. 直接法:反応障壁の算出#
分子状吸着状態から解離吸着状態へ移行する際のエネルギー障壁を、NEB(Nudged Elastic Band)法などを用いて直接算出することで、反応速度に関する知見が得られます。本基礎検討モデルは N2分子から二つのN原子への解離シミュレーションへの適用が可能です。
2. 記述子による予測法:Nørskovモデルによる相対比較#
窒素還元反応の触媒活性を評価する際、Nørskovらによる火山型触媒活性モデル [2] が広く用いられます。Advance/PHASEによる計算事例 [3] もありますが、本基礎検討の結果をそのままモデルに代入することは推奨されません。
- 誤差の相殺: 触媒活性を予測する場合、既存の計算事例 [3] のように、Ruなどの標準触媒と同一の計算設定(Primitive 2x2 スーパーセル、RPBE汎関数、窒素単原子吸着、零点振動エネルギー補正など)で比較することが重要です 。
- 系統的誤差の抑制: 設定を統一することで、溶媒効果やスラブ設定に起因する系統誤差を相殺し、精度の高い相対評価が可能となります。
まとめ#
本解析では、第一原理計算ソフトウェアAdvance/PHASEを用いて、Mo(110)表面におけるN2分子の吸着プロセスの第一段階として、解離吸着が極めて安定であることを示しました。この基礎検討の結果は、アンモニア合成における窒素供給のメカニズムを理解するための重要な指針となります。
本解析の詳細や、研究への適用可能性に関するご相談はこちら
お問い合わせ参考文献#
- E. Skulason, T. Bligaard, S. Gudmundsdóttir, F. Studt, J. Rossmeisl, F. Abild-Pedersen, T. Vegge, H. Jónssona, and J. K. Nørskov, "A theoretical evaluation of possible transition metal electro-catalysts for N2 reduction", Phys. Chem. Chem. Phys. 14, 1235 (2012).
- J. H. Montoya, C. Tsai, A. Vojvodic, and J. K. Nørskov, "The challenge of electrochemical ammonia synthesis: a new perspective on the role of nitrogen scaling relations", ChemSusChem 8, 2180 (2015).
- 電気化学的アンモニア合成における窒素還元触媒の活性・選択性の第一原理解析
関連ページ#
- 第一原理計算ソフトウェア Advance/PHASE
- 解析分野:ナノ・バイオ
- 産業分野:材料・化学