ナトリウムイオン電池向け固体電解質の探索:合成における反応エネルギーの予測と検証#

次世代の蓄電池として期待される全固体ナトリウムイオン電池の開発において、高いイオン伝導度を持ち、かつ熱力学的に安定で合成が容易な「新規固体電解質」の探索は重要な課題です。広大な組成空間から有望な候補物質を効率的に見つけ出すため、近年は機械学習による仮想スクリーニングと、第一原理計算(DFT)による物理的検証を組み合わせた「マテリアルズ・インフォマティクス(MI)」が威力を発揮しています。本事例では、有望なナトリウム系固体電解質の合成のしやすさ(熱力学的な自発性)を評価するため、「反応エネルギー」を指標としたスクリーニングを実施しました。勾配ブースティング決定木アルゴリズム「XGBoost」を用いてデータベースから反応エネルギーを高速に予測し、抽出された有望な候補物質に対して第一原理計算ソフトウェアAdvance/PHASEを用いた検証計算を行いました。機械学習モデルの予測精度と、DFT計算およびデータベースが裏付ける材料設計の妥当性について考察します。
Keywords: 第一原理計算 (DFT), マテリアルズ・インフォマティクス (MI), 全固体電池, ナトリウムイオン電池, 固体電解質, XGBoost, 反応エネルギー, PHASE-Viewer, 状態方程式 (EOS)
1. 機械学習モデルの構築とスクリーニング#
XGBoostによる生成エネルギーの予測#
XGBoost(eXtreme Gradient Boosting)は、複数の決定木を組み合わせることで高い予測精度を誇る先進的な機械学習アルゴリズムです [1]。本事例では、Materials Project(MP) [2] から約12,800件のナトリウム(Na)含有化合物のデータを取得し、物質の「生成エネルギー」を学習させました。モデルの真の予測能力を測るため、ターゲットとなる3つの候補物質(Na3PS4, Na3PSe4, Na3OBr)は学習データから意図的に除外し、ブラインドテストとしています。
化学式のみから予測を行うため、オープンソースライブラリmatminer [3] を利用し、元素ごとの基礎物理プロパティや酸化数などの統計的特徴量(Magpie特徴量等)を生成して入力としました。Optuna [4] を用いたハイパーパラメータ最適化と厳格な交差検証(GroupShuffleSplit)を行った結果、テストデータに対する予測精度は決定係数 (R2 Score): 0.96、RMSE: 0.201 eV/atomとなっています。
スクリーニング結果と特徴量の重要度#
構築したモデルを用いて、候補物質およびその原料化合物の生成エネルギーを予測し、そこから「反応エネルギー()」を算出しました。反応エネルギーが負(マイナス)に大きいほど発熱反応であり、熱力学的に合成しやすいことを示します。
表1. XGBoostによるスクリーニング結果(合成しやすい順)
| 反応式 (Reaction) | ターゲット物質 | 予測・反応エネルギー (eV/atom) |
|---|---|---|
| 1.5 Na2S + 0.5 P2S5 → Na3PS4 | Na3PS4 | -0.273 |
| 1.5 Na2Se + 0.5 P2Se5 → Na3PSe4 | Na3PSe4 | -0.127 |
| Na2O + NaBr → Na3OBr | Na3OBr | -0.015 |
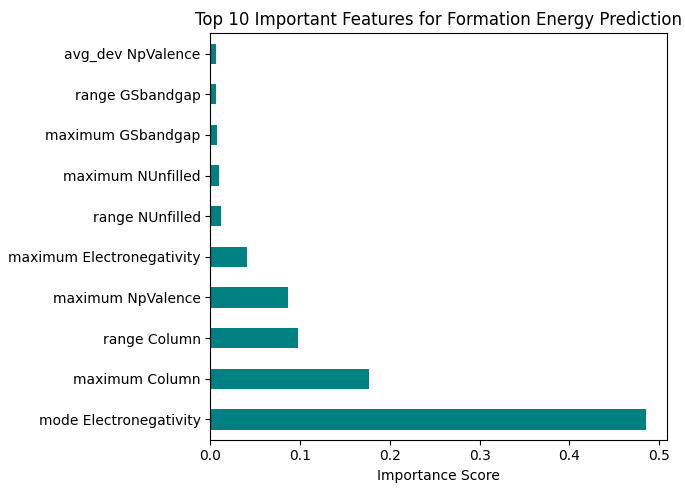
図1. 生成エネルギーの予測に寄与した特徴量の重要度(Top 10)
AI(XGBoost)の予測によれば、Na3PS4が最も合成しやすく、次いでNa3PSe4、Na3OBrと続きます。図1の特徴量重要度を見ると、モデルは「最頻(最も多く含まれる元素)の電気陰性度(mode Electronegativity)」や「周期表の族(Column)」を最重要視しています。これは、陰イオン(S, Se, O, Br等)の性質がイオン結合の強さや化合物の安定性に直結するという化学の基本原則を、AIがデータから正しく学習していることを示しています。
2. Advance/PHASEを用いたDFT計算での検証#
機械学習モデルによるスクリーニング結果の物理的妥当性を「答え合わせ」するため、第一原理計算ソフトウェアAdvance/PHASEを用いてDFT計算を実行しました。計算はPAWポテンシャルとGGA (PBE)汎関数を使用して、十分な計算条件(カットオフエネルギー、k点サンプリングなど)で行いました。
ここで、例としてターゲット物質の一つであるハライド系固体電解質候補Na3OBr(逆ペロブスカイト型構造)、およびその出発原料であるNa2O(逆蛍石型構造)とNaBr(岩塩型構造)の状態方程式(EOS: Equation of State)の計算結果を示します(図2, 図3, 図4)。それぞれ、単位胞の体積を基準値から段階的に変化させながら全エネルギーの変化を追跡し、Birch-Murnaghan状態方程式にフィッティングすることで、極小点(平衡状態)における全エネルギー()、平衡体積()、および体積弾性率()を精密に決定しました。
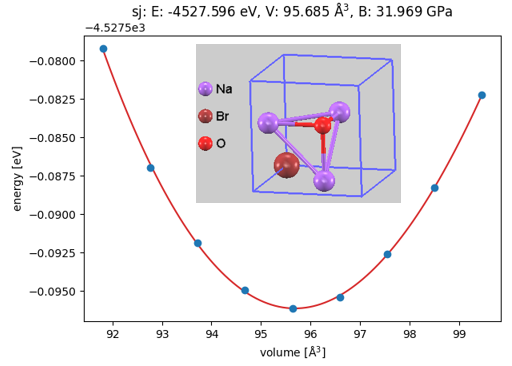
図2. Na3OBrの体積-エネルギー曲線と結晶構造
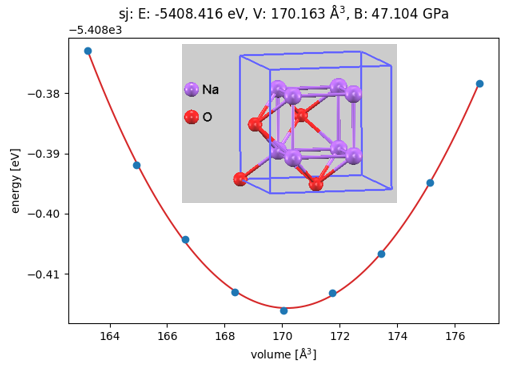
図3. Na2Oの体積-エネルギー曲線と結晶構造
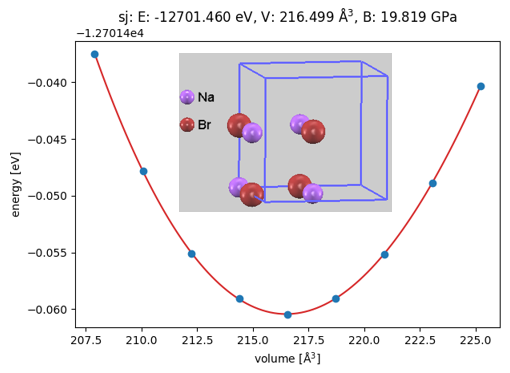
図4. NaBrの体積-エネルギー曲線と結晶構造
得られた各相の全エネルギーを基に、出発原料からNa3OBrへの反応エネルギー()を算出しました。1組成式単位(f.u.)あたりの反応エネルギーは -0.127 eV/f.u. となり、これを1原子あたりに換算すると -0.025 eV/atomとなります。このエネルギー低下(発熱)は、Na3OBrが原料の混合によって熱力学的に自発的に形成されうる、安定な相であることを裏付けています。
3. PHASE-Viewerによる反応相図の取得とクロスバリデーション#
Advance/PHASEのGUIであるPHASE-Viewerには、第一原理計算の入力作成・計算実行・結果表示だけでなく、外部材料データベースであるMaterials Projectとシームレスに連携する機能が備わっており、熱力学的な安定性を可視化する「反応相図(Reaction Diagram)」をダイレクトに検索・生成することが可能です。
図5. PHASE-Viewerにおける反応相図の検索入力画面。出発原料(Na2O, NaBr)を指定することで直ちに検索可能。
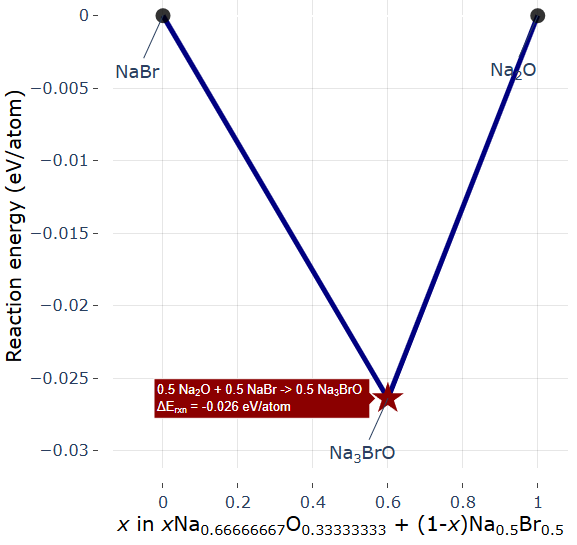
図6. 検索によって得られたNaBr-Na2O擬二元系における反応相図(凸包)。V字の谷の最深部に安定相であるNa3OBrがプロットされています。
図5に示すPHASE-Viewerのインターフェースから原料化合物を指定して検索を実行した結果、図6の反応相図(エネルギー凸包図)が取得されました。この相図は、NaBr(左端)とNa2O(右端)の混合比()を変化させた際の反応エネルギーを示しており、(Na3OBr組成)において明確な「V字の谷(赤い星マーク)」を形成しています。これは、この組成において混合系が最も安定化すること、すなわち未知の競合相に分解することなく単相として合成可能であることを明瞭に示しています。Materials Projectに登録されている参照データでは、この谷の深さ(反応エネルギー)は-0.026 eV/atomと記録されています。
ここで、本事例で用いた各アプローチ(機械学習予測、Advance/PHASEによる独自DFT計算、Materials Projectデータベース値)による反応エネルギーの算出結果を一覧にまとめ、多角的なクロスバリデーション(相互検証)を行いました。
表2. 各手法による反応エネルギー( [eV/atom])の比較
| ターゲット物質 | XGBoost 予測値 (MI) | Advance/PHASE 計算値 (DFT) | Materials Project 参照値 (DB) |
|---|---|---|---|
| Na3PS4 | -0.273 | -0.208 | -0.208 |
| Na3PSe4 | -0.127 | -0.144 | -0.144 |
| Na3OBr | -0.015 | -0.025 | -0.026 |
予測と検証に関する詳細な考察#
表2の比較から興味深い知見が確認できます。
第一に、Advance/PHASEを用いた第一原理計算値が、Materials Projectのデータベース値と高精度に一致している点です。Materials Projectでは生成エネルギーの計算に単体(気相)やアニオンなどのエネルギーに対して独自の補正(correction)を組み込んでいるため、生成エネルギーの比較には特に注意が必要です。一方、今回の固体電解質の合成反応はすべて固体相間の反応であり、反応前後でのエネルギー差をとる過程で単体基準の補正値が相殺されるため、基準の違いによる問題が解消されています。また、計算条件を適切に設定すれば、汎関数や擬ポテンシャルの影響(系統誤差)が相殺され、高精度な計算が実現できることが示されています。
第二に、候補物質を学習データから除外し、未知のデータとして推論を行わせたXGBoostモデルが、「Na3PS4 > Na3PSe4 > Na3OBr」という合成のしやすさ(発熱エネルギーの大きさ)の序列を正しく予測できている点です。絶対値にはわずかなデータ駆動型特有の滑らかさ(平均への回帰)による誤差が見られますが、広大な組成空間からアタリを付ける仮想スクリーニングツールとして、機械学習が今後の固体電解質の探索に大きなポテンシャルを示しています。
第三に、本モデルのテストデータ(生成エネルギー)に対する予測精度は決定係数(R2 Score)が0.96と高い一方、RMSEは0.201 eV/atomに達している点です。数 meV〜数十 meV/atom の微小な反応エネルギーを議論する材料科学において、この誤差は一見すると実用に耐えないほど過大に思われます。しかし、本スクリーニングが実効的に機能した背景には、データ駆動型アプローチにおける「誤差の相殺」と「相対評価の有用性」があります。
まず、ターゲット指標を物質の生成エネルギーではなく、その差分である「反応エネルギー()」としたことで、右辺(生成物)と左辺(原料)で原子の種類と数が完全に一致し、モデルが抱える元素ごとの系統誤差が引き算の過程で都合よく相殺されたと考えられます(これは第一原理計算において系統誤差が相殺される性質とも類似しています)。また、本MIワークフローの目的は絶対値の厳密な的中ではなく、有望な候補を絞り込むための「序列(トレンド)の評価」です。陰イオンの性質に起因する化学的な安定性の序列さえ正しく捉えられていれば、一次スクリーニングとして有用です。さらに、今回の対象物質の真の反応エネルギー(シグナル)が比較的大きく、ノイズに埋もれなかったことも有利に働きました。このように、機械学習モデル単体の限界(誤差)を正しく認識した上で、精度検証を担う第一原理計算へと繋ぐ「両輪(二段構え)」のワークフローを構築することの重要性が、本事例から明確に示されています。
まとめ#
本事例では、マテリアルズ・インフォマティクスにおける実用的な二段構えの材料探索ワークフローを実証しました。データベースとXGBoostを用いたマクロな「高速仮想スクリーニング」によって有望な固体電解質候補の序列を絞り込み、Advance/PHASEによるミクロな「高精度第一原理検証」によって熱力学的な安定性を裏付けました。データ科学による高速推論と、計算科学による精度検証という「両輪」をシームレスに連携させ、最終的な実験による合成実証へと繋げることで、次世代固体電解質材料開発のコストと時間を大きく削減することが可能です。
本解析の詳細や、研究への適用可能性に関するご相談はこちら
お問い合わせ参考文献#
- T. Chen and C. Guestrin, "XGBoost: A Scalable Tree Boosting System", Proceedings of the 22nd ACM SIGKDD International Conference, 785 (2016).
- A. Jain et al., "Commentary: The Materials Project: A materials genome approach to accelerating materials innovation", APL Materials 1, 011002 (2013).
- L. Ward et al., "Matminer: An open source toolkit for materials data mining", Comput. Mater. Sci. 152, 60 (2018).
- T. Akiba, S. Sano, T. Yanase, T. Ohta, and M. Koyama, "Optuna: A Next-generation Hyperparameter Optimization Framework", Proceedings of the 25th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, 2623 (2019).
関連ページ#
- 第一原理計算ソフトウェア Advance/PHASE
- 解析分野:ナノ・バイオ
- 産業分野:材料・化学