データ駆動型アプローチによるNa系固体電解質の密度予測と第一原理計算による検証#

次世代の蓄電池として期待される全固体ナトリウムイオン電池の開発において、高いイオン伝導度を持ち、かつ構造的に安定な「新規固体電解質」の探索は重要な課題です。物質の基本的な物理量である「密度」は、セル設計におけるエネルギー密度や、結晶格子内のイオンの動きやすさ(隙間の多さ)に直結する重要な指標となります。広大な組成空間から有望な候補物質を効率的に見つけ出すため、本事例では機械学習(XGBoost)を用いた仮想スクリーニングと、第一原理計算ソフトウェアAdvance/PHASEを用いた状態方程式(EOS)による物理的検証を組み合わせた「マテリアルズ・インフォマティクス(MI)」のアプローチをご紹介します。
Keywords: 第一原理計算 (DFT), マテリアルズ・インフォマティクス (MI), 全固体電池, ナトリウムイオン電池, 固体電解質, XGBoost, 密度予測, 状態方程式 (EOS)
1. 機械学習モデルの構築とスクリーニング#
XGBoostによるNa系固体電解質の密度予測#
XGBoost(eXtreme Gradient Boosting)[1] は、複数の決定木を直列に繋いで学習を繰り返す「勾配ブースティング」と呼ばれる強力なアンサンブル学習アルゴリズムです。特徴量間の複雑な非線形関係を自動的に捉える能力に優れており、マテリアルズ・インフォマティクスにおける物性予測でも広く活用されています。
本事例では、Materials Project(MP)[2] から約8,000件のナトリウム(Na)を含有する非金属化合物のデータを取得し、物質の「密度」を学習させました。モデルの汎化性能を評価するため、ターゲットとなる3つの候補物質系(Na3BrO, Na3PS4, Na3PSe4)は学習データから意図的に除外し、未知の化学系に対するブラインドテストを実施しています。
特徴量にはmatminer [3] を利用し、MagpieやMeredig、電気陰性度の差分といった組成ベースの記述子に加え、物理法則(平均原子量を共有結合半径の3乗で割った擬似密度など)に基づくドメイン知識を注入しました。Optuna [4] によるハイパーパラメータ最適化と厳格な交差検証を行った結果、未知データに対する予測性能として決定係数 (R2 Score): 0.909、RMSE: 0.282 g/cm3 という精度を達成しました。
図1は、テストデータに対する実測密度とXGBoostによる予測密度の関係を示すパリティプロットです。全体的な傾向としてデータ点は理想線(y = x、赤色の破線)の周囲に分布しており、広大な探索空間からの大まかな密度予測(スクリーニング)が可能であることが確認できます。一方で、一部のデータ点が理想線から外れてばらつく傾向も見られます。その主な原因は、組成の変化に対して、最安定となる結晶構造のタイプ(原子の詰まり具合)が非連続に変化することにあります。組成記述子のみを入力とするXGBoostモデルは、この結晶構造の変化を完全に予測することには限界があります。このスクリーニング能力の有用性と予測限界の存在こそが、後段で実施する第一原理計算による検証の重要性を際立たせています。
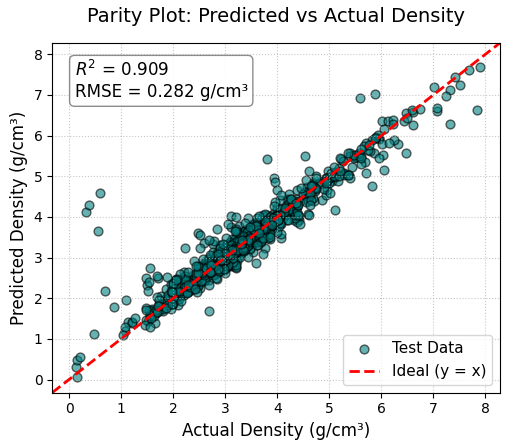
図1. テストデータに対する実測密度と予測密度のパリティプロット。低密度から高密度まで、理想線(赤破線)に沿った安定した予測ができていることが分かります。
特徴量の重要度#
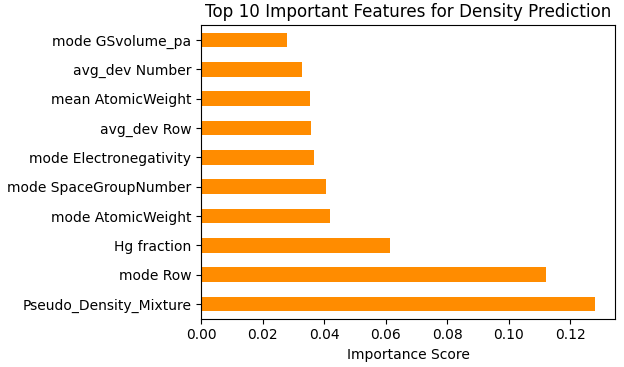
図2. 密度予測に寄与した特徴量の重要度(Top 10)
図2の特徴量重要度を見ると、本解析で導入したドメイン特徴量である「単体を混合したと仮定した際の理論密度(Pseudo_Density_Mixture)」が最も高く寄与しています。これは、アルゴリズム単体の学習能力だけでなく、物理法則(質量と体積の関係)に基づく適切な特徴量設計(人間のドメイン知識)を与えることが、高精度な推論モデルの構築において重要であることを示唆しています。機械学習モデルは、この古典的な近似式を強力なベースラインとしつつ、他の多様な特徴量と組み合わせて非線形な補正を加える役割を果たしています。
2. Advance/PHASEを用いたDFT計算での検証#
機械学習モデルによるスクリーニング結果の物理的妥当性を検証するため、Advance/PHASEを用いてDFT計算 (PAWポテンシャル/GGA-PBE汎関数利用)を実行しました。ターゲット物質の結晶構造モデル(Na3BrO, Na3PS4, Na3PSe4)に対し、単位胞の体積を段階的に変化させながら全エネルギーを計算し、Birch-Murnaghan状態方程式にフィッティングすることで平衡体積を求めました。
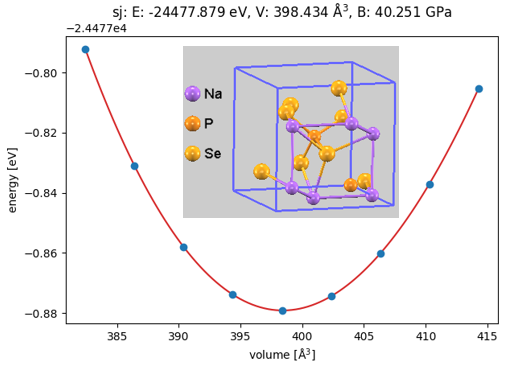
図3. Na3PSe4(慣用単位胞:Na6P2Se8)の体積-エネルギー曲線と結晶構造。V=398.434 Å3でエネルギーが極小となる平衡状態を示しています。
図3に示すように、状態方程式から平衡体積を求め、これを用いて密度を算出しました。この密度のDFT計算値とXGBoostによる推論値、およびMaterials Projectの参照値とのクロスバリデーション(相互検証)を行いました。
3. 予測結果と第一原理計算のクロスバリデーション#
表1. 各手法による理論密度 [g/cm3] の比較
| ターゲット物質 | XGBoost 予測値 (MI) | Advance/PHASE 計算値 (DFT) | Materials Project 参照値 (DB) |
|---|---|---|---|
| Na3BrO | 2.897 | 2.861 | 2.939 |
| Na3PS4 | 2.160 | 2.182 | 2.234 |
| Na3PSe4 | 3.401 | 3.466 | 3.588 |
予測と検証に関する詳細な考察#
表1の比較結果から、マテリアルズ・インフォマティクスの有効性を示す興味深い知見が得られました。
第一に、候補物質を学習データから除外したブラインド予測における、機械学習モデルの本質的な性能評価についてです。注目すべきは、ターゲット物質の密度における「大小関係の序列(トレンド)」を、組成情報のみから正しく再現できている点にあります。これは、機械学習モデルが個別の物質に対する厳密な絶対値の予測には限界を残す半面、広大な組成空間から有望な候補の傾向を正しく捉えるスクリーニングツールとして、実用的な精度を有していることを示しています。
第二に、Advance/PHASE(GGA-PBE)による計算値は、Materials Project(r²SCAN/PBEsol [5])の参照値と比較して全体的にわずかに密度が低く(体積が大きく)算出される系統的な傾向が見られます。これは、PBE汎関数が格子定数をわずかに過大評価する傾向があるのに対し、PBEsol汎関数が固体の格子定数をより高精度に予測するように作成されたPBEの変種であるという、汎関数の性能に関する一般的な傾向を明確に反映しています。
まとめ#
本事例では、マテリアルズ・インフォマティクスにおける実用的な二段構えの材料探索ワークフローを実証しました。データベースとXGBoostを用いた「高速仮想スクリーニング(密度予測)」によって膨大な候補から有望な物質群の序列を大まかに絞り込み、Advance/PHASEによる「第一原理検証(平衡体積算出)」によってその物理的な妥当性を確認・補正しています。第一原理計算における汎関数の特性(系統的な誤差)をあらかじめ把握しておく必要はありますが、データ科学による広範な探索と計算科学による詳細な検証という「両輪」を連携させることで、次世代材料開発のコストと時間を効果的に削減することが期待できます。
本解析の詳細や、研究への適用可能性に関するご相談はこちら
お問い合わせ参考文献#
- T. Chen and C. Guestrin, "XGBoost: A Scalable Tree Boosting System", Proceedings of the 22nd ACM SIGKDD International Conference, 785 (2016).
- A. Jain et al., "Commentary: The Materials Project: A materials genome approach to accelerating materials innovation", APL Materials 1, 011002 (2013).
- L. Ward et al., "Matminer: An open source toolkit for materials data mining", Comput. Mater. Sci. 152, 60 (2018).
- T. Akiba, S. Sano, T. Yanase, T. Ohta, and M. Koyama, "Optuna: A Next-generation Hyperparameter Optimization Framework", Proceedings of the 25th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, 2623 (2019).
- R. Kingsbury, A. S. Gupta, C. J. Bartel, J. M. Munro, S. Dwaraknath, M. Horton, and K. A. Persson, "Performance comparison of r²SCAN and SCAN metaGGA density functionals for solid materials via an automated, high-throughput computational workflow", Phys. Rev. Materials 6, 013801 (2022).
関連ページ#
- 第一原理計算ソフトウェア Advance/PHASE
- 解析分野:ナノ・バイオ
- 産業分野:材料・化学