Partial Hessian法によるフォノン計算の高速化と吸着自由エネルギーの算出#

表面への分子吸着は、触媒反応やガスセンサーなどの機能発現における第一歩です。吸着状態の熱力学的な安定性を正確に評価するためには、純粋な電子エネルギー()だけでなく、温度効果やゼロ点振動エネルギー、エントロピーを含んだ「吸着自由エネルギー()」の算出が不可欠です。しかし、金属スラブモデルの全原子を対象としたフォノン(振動)計算は、多大な計算コストを要するだけでなく、スラブモデル特有の低周波モードの混入によるエントロピーの発散という計算化学的な課題(罠)を抱えています。本事例では、第一原理計算ソフトウェアAdvance/PHASEを用い、振動させる原子を限定する「Partial Hessian(部分ヘシアン)法」を活用することで、計算コストを大幅に削減しつつ安定的にPd(111)表面上のCO吸着自由エネルギーを算出する実践的なワークフローを紹介します。
Keywords: 第一原理計算, Partial Hessian法, フォノン計算, 吸着自由エネルギー, Pd(111), CO, 振動解析, Cut-off法
計算モデルと計算条件#
本解析では、Pd(111)表面を模擬した3×3のスーパーセルモデル(4層スラブモデル)を作成し、その上に1分子のCOを配置しました。表面セルの格子定数は最適化されたバルクの格子定数に基づいています。カバレッジを1/9 ML(モノレイヤー)の低カバレッジに設定することで、隣接するCO分子間の双極子-双極子相互作用による人工的な振動数シフトを排除し、孤立分子としての純粋な吸着状態を評価しています。計算コストとスラブのバルク的性質を両立させるため、下部2層のPd原子は固定(Fix)して計算を行いました。
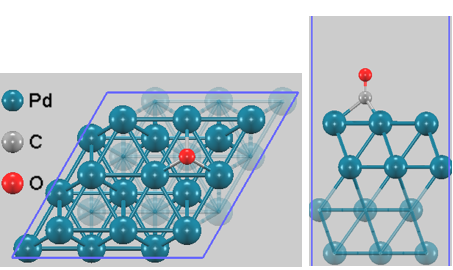
図1. Pd(111) 3×3セル上のCO吸着モデル(fcc中空サイト)。下部2層(半透明表示)は固定して計算を実行。
表1. 計算条件の概要
| 項目 | 設定 |
|---|---|
| 擬ポテンシャル | ウルトラソフト擬ポテンシャル |
| 交換相関汎関数 | GGA (PBE) |
| van der Waals補正 | DFT-D3 (Grimme法) |
| カットオフエネルギー | 25 Rydberg |
| k点サンプリング | 4×4×1(スラブモデル) / 1×1×1(点:気相孤立CO分子) |
| 有限変位幅 (Finite displacement) | 0.025 bohr |
Partial Hessian法の設定とアルゴリズムの特長#
Advance/PHASEのGUI環境では、Partial Hessian計算を直感的に設定することが可能です。図2に示すように、力計算の対象とする原子範囲を、原子の「分子(グループ)属性」から自動的に抽出(auto-define)し指定することができます。これにより、原子数の多い複雑なスラブモデルであっても、手動で原子インデックスを探す手間が省け、人為的な設定ミスを防ぐことができます。
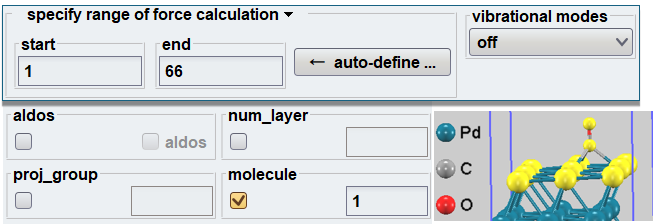
図2. GUIを用いたPartial Hessian計算の対象原子指定。選択された原子(黄色)の分子属性から力計算の範囲を自動設定可能。
【マトリックス対角化におけるアルゴリズム】
一般的な第一原理計算コードによる部分ヘシアン計算では、系全体の枠組み(全原子数 )を維持したまま、計算対象外の基板原子の力の定数を「ゼロ」として扱います。このとき対角化される動的行列(Dynamical matrix) は以下のようになります。
ここで、 は吸着種のヘシアン行列、 は吸着種と基板の結合項(非対角項)です。基板部分は として扱われますが、結合項が残存しているため、この行列を対角化すると数学的に必ず「正の解(吸着種の物理的な振動)」と「負の解(基板の不安定モード)」がペアで出力されてしまいます。この大量の大きな負の振動数(アーティファクト)が結果ファイルに混在するため、物理的に正しいモード(正の振動数のモード)の抽出に注意が必要となります。
一方、Advance/PHASEで推奨される手法では、力計算を行った後に force.data のファイルをreduce_force_dataツールで編集し、さらに対象外の基板原子を削除してから対角化を実行します。処理される行列 は以下の通りです。
次元が に縮小されているため、アーティファクトとしての負の振動数は原理的に発生せず、物理的に妥当な正の振動数のみを出力させることができます。
計算結果と考察#
1. 可動原子範囲の比較と「分子寄与率フィルタリング」#
構造最適化により、CO分子はPd(111)表面のfcc中空サイトに最安定吸着することが確認されました。スラブモデルのような系において、全原子を変位させるFull Hessian計算はコストを要します。そこで、前述の特長を活かし、動かす原子を①「CO分子のみ(2原子)」に限定したモデルと、②「CO分子+最表面のPd層(11原子)」に拡大したモデルの2パターンでPartial Hessian計算を実行し、得られた振動数と安定性を比較しました。
ここで重要になるのが、熱力学量を評価するための「分子寄与率フィルタリング」です。本解析に用いたPythonスクリプトでは、出力された全ての振動モードの中から、「吸着分子(C, O)が一定割合(例:30%)以上動いているモード」だけを抽出し、金属スラブ自身のフォノンモードを自動的に除外します。
表2. 各モデルにおけるC-O内部伸縮振動数の比較 (単位: cm-1)
| モードの種類 | CO分子のみ 可動 (2原子) | CO+第1層Pd 可動 (11原子) | 実験値 [1] |
|---|---|---|---|
| C-O内部伸縮 | 1773.0 | 1800.4 | 1823 |
考察:
- 高周波モードの高精度な再現: C-O伸縮振動は、どちらのモデルでも実験的に観測される中空サイト特有の帯域 [1] をよく再現しています。「COのみ」に限定しても実用上十分な精度が得られており、計算コストの削減効果と物理的妥当性が確認できます。
- 低周波モードの混在: 表2には記載していませんが、並進や回転の束縛に由来する低周波モードの振る舞いはモデルによる違いが現れます。「COのみ」モデルでは剛体表面に対する揺れとして241 cm-1付近に明確に現れますが、「CO+第1層Pd」モデルではPd-C結合のバネが連動するため、約190〜365 cm-1 付近に複数のモードとして混在します。
- スラブ計算の「罠」: 「CO+第1層Pd」を動かしたモデルでは、Pd-C間の結合バネがよりリアルに表現される一方、非常に小さい振動数を持つモードが多数現れました。これらのモードは後述のように自由エネルギーの計算に大きな問題を引き起こします。
2. 振動自由エネルギーの補正と「Cut-off法」#
振動解析から得られた各モードの周波数を用いて、系のゼロ点振動エネルギー(ZPE)および温度・エントロピーの寄与(298.15 K)を算出します。しかし、数十 cm-1 以下の極めて低い振動モード(並進や回転の束縛モード)を調和振動子近似の式に代入すると、振動数が0に近づくにつれてエントロピー項(-TS)が無限大へ発散してしまう問題が生じます [2]。
本事例ではこの問題の実用的な回避策として、Pythonスクリプトを用いて「100 cm-1 以下の低周波モード(および計算ノイズによる微小な虚数振動)を一律に 100 cm-1 に切り上げて扱う Cut-off法」を適用し、安定した熱力学補正項 を算出しました。
- (COのみ可動モデル, Cut-off適用) = 0.065 eV
- (CO+第1層Pd 可動モデル, Cut-off適用) = 0.160 eV
3. 吸着自由エネルギー (ΔGads) の算出#
CO+第1層Pd 可動モデルにおいて、以下の熱力学サイクルに基づく関係式から吸着自由エネルギーを求めます。
ここで、 はDFT構造最適化によって得られた純粋な電子エネルギーの差分です。また、気相中の孤立CO分子の補正項 には、理想気体近似とNISTの実験データベースに基づく定数(-0.386 eV)を採用しました [3]。なお、吸着前のPdスラブ単体の振動寄与については、前述の分子寄与率フィルタリングによってスラブのみのフォノンモードが除外されるため、本サイクルにおいては考慮不要(ゼロ)となります。
表3. 吸着自由エネルギーの算出内訳 (298.15 K, 1 atm)
| 項目 | エネルギー値 (eV) |
|---|---|
| (純粋な電子吸着エネルギー) | -2.254 |
| (吸着状態の熱力学補正) | +0.160 |
| (気相COの熱力学補正) | -0.386 |
| (吸着自由エネルギー) | -1.708 |
最終的な吸着自由エネルギーとして-1.708 eV が得られました。純粋な電子エネルギー(-2.254 eV)と比較すると、自由エネルギーは約0.55 eVほどプラス側へシフト(不安定化)しています。これは、気相中で自由に飛び回っていたCO分子が表面に束縛されることで、大きなエントロピーペナルティ(並進・回転の自由度の喪失)を受けたことを物理的に正しく反映した結果です。
なお、文献 [4] で指摘しているように、PBEなどの半局所密度汎関数は、遷移金属表面へのCO吸着において、金属から分子への電荷移動の誤りに起因する「密度主導の誤差(density-driven errors)」を抱えており、吸着エネルギー()を過大評価(Overbinding)してしまう傾向 [5] があります。また、計算で得られる絶対的なエネルギー値は使用する汎関数に大きく依存します。したがって、実験結果と比較する際には、温度効果によるエントロピー変化など、熱力学的な相対シフトに着目して評価することがより適切と言えます。
まとめ#
本解析では、第一原理計算ソフトウェアAdvance/PHASEで実装されたPartial Hessian法を用いて、Pd(111)表面上のCO吸着系におけるフォノン計算コストを大幅に削減しつつ、温度・エントロピー効果を適切に反映した吸着自由エネルギーを算出しました。振動させる原子を「吸着分子とその周辺」に限定するアプローチと、行列を物理的に縮小して不要なアーティファクトを排除するアルゴリズム、さらにはエントロピー発散を防ぐCut-off法を併用することで、金属表面系特有の計算化学的な「罠」を回避できることが実証されました。これらの知見と機能は、より複雑な触媒反応ネットワークの構築や、ハイスループットな吸着状態スクリーニングにおいて強力なツールとなります。
本解析の詳細や、研究への適用可能性に関するご相談はこちら
お問い合わせ参考文献#
- A. M. Bradshaw and F. M. Hoffmann, "The chemisorption of carbon monoxide on palladium single crystal surfaces: IR spectroscopic evidence for localised site adsorption", Surf. Sci. 72, 513-535 (1978).
- S. Grimme, "Supramolecular Binding Thermodynamics by Dispersion-Corrected Density Functional Theory", Chem. Eur. J. 18, 9955-9964 (2012).
- M. W. Chase Jr., "NIST-JANAF Thermochemical Tables, 4th Edition", J. Phys. Chem. Ref. Data, Monograph 9 (1998); NIST Chemistry WebBook, SRD 69.
- A. Patra, H. Peng, J. Sun, and J. P. Perdew, "Rethinking CO adsorption on transition-metal surfaces: Effect of density-driven self-interaction errors", Phys. Rev. B 100, 035442 (2019).
- M. Gajdoš, A. Eichler, and J. Hafner, "CO adsorption on close-packed transition and noble metal surfaces: trends from ab initio calculations", J. Phys. Condens. Matt. 16, 1141 (2004).
関連ページ#
- 第一原理計算ソフトウェア Advance/PHASE
- 解析分野:ナノ・バイオ
- 産業分野:材料・化学