時間依存密度汎関数法(TDDFT)を用いた誘電関数スペクトル・光吸収断面積の計算事例#

半導体デバイスの受光感度や、有機エレクトロニクス材料の発光特性を高精度に予測するためには、電子の励起状態を正しく記述できる理論が必要です。時間依存密度汎関数法(TDDFT)は、計算コストと精度のバランスに優れた励起状態計算手法として、産業界でも導入が進んでいます。本解析では、第一原理計算ソフトウェアAdvance/PHASEのTDDFT機能を用い、Si結晶の誘電関数スペクトルおよびベンゼン分子の光吸収断面積を計算した事例を紹介します。
Keywords: 第一原理計算, TDDFT, 誘電関数, 光吸収, 線形応答 (linear response)
理論的背景#
通常の密度汎関数法(DFT)が基底状態の電子密度を扱うのに対し、TDDFTは時間依存する外部ポテンシャル 下での電子密度の時間発展 を記述します。Runge-Grossの定理により、時間依存する電子密度は一意に外部ポテンシャルを決定するため、系は以下の時間依存Kohn-Sham方程式に従います。
ここで、有効ポテンシャル には、外部ポテンシャル、Hartree項に加え、時間依存する交換相関ポテンシャル が含まれます。この基礎方程式からスペクトルを得るには、大きく分けて以下の2つのアプローチが存在します。
(1) Real-time TDDFT (実時間発展法)#
Yabana-Bertschスキーム [1]やSugino-Miyamotoメソッド [2] などが代表的です。系に対し、時刻 でパルス電場などの摂動をあらわに加えます。その後、時間依存Kohn-Sham方程式に従って波動関数を実時間でステップごとに発展させます。この時間発展中の電子密度の変化から双極子モーメント を計算し、これをフーリエ変換することで周波数依存の分極率や光吸収スペクトルを直接得ます。また、強い外場による非線形光学特性やTDDFT-MDシミュレーション [3]への展開もなされています。
(2) Frequency-domain TDDFT (周波数領域法)#
光応答のような摂動が十分に弱いと仮定し、TDDFTの計算式に対してのみ摂動論を適用して線形化(Linearization)を行います。これを時間領域から周波数領域へフーリエ変換し、線形応答方程式として定式化して解く手法です。Advance/PHASEでは、この周波数領域のアプローチを採用し、外部摂動に対する密度応答関数(感受率) を求めるため、以下のDyson型の方程式(Dyson-like equation)を用いています [4]。
あるいは、これを変形して連立一次方程式の形にした以下の式を用います。
ここで、 は非相互作用系(Kohn-Sham系)の応答関数、 はクーロン相互作用、 は交換相関カーネルです。Advance/PHASEでは、この方程式を逆格子空間(G空間)で表現し、各エネルギー点 ごとに行列演算(連立方程式の解法)を行うことで誘電関数スペクトルを算出します。この手法は結晶などの周期系に適しています。
また、分子系などの孤立系に対しては、遷移空間(電子-正孔対基底)を用いたBethe-Salpeter様方程式(BS-like equation)に基づくソルバー [5] も実装されています。これは基底のとり方が異なるのみで、物理的には等価な線形応答方程式を解いています。
Casida方程式との違い
周波数領域TDDFTの標準的な実装として知られるCasida方程式 [5]は、励起エネルギー を直接求めるための固有値問題()として定式化されます。これに対し、本手法(Dyson/BSソルバー)は、特定の周波数 における応答関数の値を連立一次方程式として解くアプローチをとります。このため、広範囲の連続スペクトル(誘電関数スペクトル全体など)を効率的に計算するのに適しています。
解析結果#
Si結晶の誘電スペクトル#
図1は、Si結晶の誘電関数スペクトル(虚部)の計算結果です。青線は独立粒子近似(RPA: Random Phase Approximation、)の結果、赤線は長距離相互作用の補正(LRC: Long-Range Correction)[6] を取り入れたTDDFTの結果を示しています。
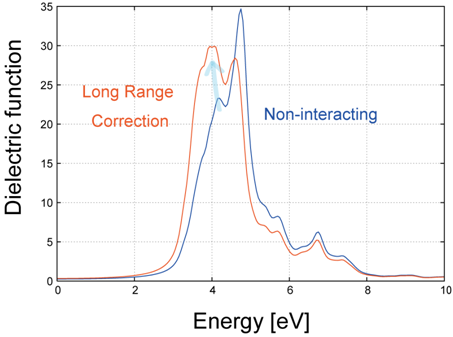
図1. LRCカーネルを用いたSi結晶の誘電スペクトル(虚部)。青線:独立粒子近似、赤線:TDDFT (LRC)。矢印はTDDFT導入による第1ピークの強度の増大を示します。
独立粒子近似と比較して、TDDFT(LRC)では低エネルギー側(約4 eV付近)の第1ピークの強度が顕著に増大していることがわかります。これは電子と正孔の引力的な相互作用(励起子効果の一部)が取り込まれた結果です。なお、本計算ではDFT共通の課題であるバンドギャップの過小評価自体は改善されていませんが、TDDFTを用いることで、独立粒子近似では再現できない「実験スペクトルに近いピーク形状や強度比」を定性的に再現できており、光デバイス材料の光学特性を相対評価する上で有用な指針を与えます。
C6H6分子の光吸収断面積#
図2は、ベンゼン(C6H6)分子の光吸収断面積スペクトルの計算結果です。ここでは断熱局所密度近似(ALDA)カーネルを使用しています。
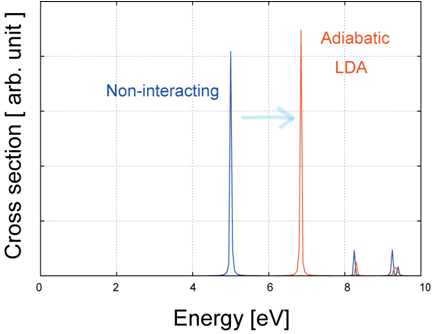
図2. ALDAカーネルを用いたC6H6分子の光吸収断面積。青線:独立粒子近似、赤線:TDDFT (ALDA)。
独立粒子近似(青線)と比較して、TDDFT(赤線)では吸収ピークが高エネルギー側(ブルーシフト)に移動していることが確認できます。これは、分子のような有限系において、Hartree項や交換相関カーネルによる多体効果の補正(電子・正孔相互作用など)が励起エネルギーに寄与し、単一粒子近似では不十分だった励起状態の記述をより高精度化させているためです。これにより、実験結果とより整合する励起スペクトルが得られます。
今後の展望:非断熱ダイナミクスによる劣化・反応解析#
TDDFTの応用は特定の構造における静的なスペクトル計算(励起エネルギー・振動子強度)にとどまりません。近年、光励起後の化学反応や超高速緩和過程をシミュレーションする非断熱分子動力学(Non-adiabatic Molecular Dynamics)への関心が高まっています。これは、Born-Oppenheimer近似が破綻する励起状態間での遷移(ホッピング)を伴う原子核の運動を記述するものです。
こうしたシミュレーションには、SHARC(Surface Hopping including ARbitrary Couplings)[7] などの公開コードが利用可能になってきています。TDDFTは、CASSCFやCASPT2などの多体波動関数法に比べて計算コストが大幅に抑えられるため、膨大な数の構造サンプリングを必要とする非断熱ダイナミクスのためのポテンシャル曲面生成手法として有力です。また、TDDFTの線形応答形式から非断熱結合係数(Non-Adiabatic Couplings: NACs)を効率的に計算する定式化が提案され [8, 9]、非断熱ダイナミクスへの応用が進んでいます。
これにより、TDDFTを用いた第一原理計算ベースでの光化学反応シミュレーションが可能となりつつあり、太陽電池材料や光触媒の電荷分離プロセスの効率化や、ディスプレイ材料の光劣化メカニズムの解明といった、産業界が抱える「効率」と「寿命」に関わる課題解決への道を開くものです。今後ますますの応用拡大が期待されます。
まとめ#
本事例では、第一原理計算ソフトウェアAdvance/PHASEのTDDFT機能(線形応答理論に基づくDyson/BSソルバー)を用いて、Si結晶の誘電関数とベンゼン分子の光吸収断面積を計算しました。G空間または遷移空間での連立一次方程式解法を採用することで、広帯域のスペクトルを高効率に取得できることを確認しました。また、LRCやALDAといった交換相関カーネルを適切に選択することで、独立粒子近似では再現できない励起子効果やスペクトルのシフトを記述でき、実験結果とより整合する光学特性解析が可能であることが示されました。
本解析の詳細や、研究への適用可能性に関するご相談はこちら
お問い合わせ参考文献#
- K. Yabana and G. F. Bertsch, "Time-dependent local-density approximation in real time", Phys. Rev. B 54, 4484 (1996).
- O. Sugino and Y. Miyamoto, "Density-functional approach to electron dynamics: Stable simulation under a self-consistent field", Phys. Rev. B 59, 2579 (1999).
- J. Haruyama, C. Hu, and K. Watanabe, "First-principles molecular-dynamics simulation of biphenyl under strong laser pulses by time-dependent density-functional theory", Phys. Rev. A 85, 062511 (2012).
- G. Onida, L. Reining, and A. Rubio, "Electronic excitations: density-functional versus many-body Green’s-function approaches", Rev. Mod. Phys. 74, 601 (2002).
- M. E. Casida, "Time-Dependent Density Functional Response Theory for Molecules", in Recent Advances in Density Functional Methods, Part I, edited by D. P. Chong (World Scientific, Singapore, 1995), p. 155.
- S. Botti, F. Sottile, N. Vast, V. Olevano, L. Reining, H.-C. Weissker, A. Rubio, G. Onida, R. Del Sole, and R. W. Godby, "Long-range contribution to the exchange-correlation kernel of time-dependent density functional theory", Phys. Rev. B 69, 155112 (2004).
- S. Mai, P. Marquetand, and L. González, "Nonadiabatic dynamics: The SHARC approach", WIREs Comput. Mol. Sci. 8, e1370 (2018).
- C. Hu, H. Hirai, and O. Sugino, "Nonadiabatic couplings from time-dependent density functional theory: Formulation in the Casida formalism and practical scheme within modified linear response", J. Chem. Phys. 127, 064103 (2007).
- C. Hu, O. Sugino, H. Hirai, Y. Tateyama, "Nonadiabatic couplings from the Kohn-Sham derivative matrix: Formulation by time-dependent density-functional theory and evaluation in the pseudopotential framework", Phys. Rev. A 82, 062508 (2010).
関連ページ#
- 第一原理計算ソフトウェア Advance/PHASE
- 解析分野:ナノ・バイオ
- 産業分野:材料・化学