積層欠陥エネルギーの第一原理計算:ANNNIモデルによる4H/6H-SiCの評価#

炭化ケイ素(SiC)は、高耐圧・低損失なパワー半導体材料として広く実用化されています。SiCは同じ組成でありながら積層順序の異なる多数の「ポリタイプ」をもち、なかでも4H-SiCと6H-SiCが代表的です。これらの結晶では基底面(0001)上の「積層欠陥(stacking fault)」が転位の分解幅や、パワーデバイスの通電劣化(バイポーラ劣化)に直結するため、その生成しやすさを支配する積層欠陥エネルギー(SFE)を正確に知ることは、材料・デバイス設計において重要です。しかしSiCのSFEは他の四面体配位半導体より桁違いに小さく(数mJ/m2)、積層欠陥を含む大規模スーパーセルの直接計算では精度の確保が容易ではありません。本解析では、第一原理計算ソフトウェアAdvance/PHASEを用い、少数の小さなポリタイプの全エネルギーからANNNI(軸性次近接イジング)モデルを介して4H-SiCと6H-SiCのSFEを効率的かつ高精度に評価します。
Keywords: 第一原理計算, DFTシミュレーション, 積層欠陥エネルギー, ANNNIモデル, 炭化ケイ素(SiC), ポリタイプ, 層間相互作用
計算方法:ANNNIモデルによる積層欠陥エネルギーの算出#
積層欠陥は「ポリタイプの極限構造」とみなせます。この考えに基づき、いくつかの小さなポリタイプの全エネルギーを層間(二重層間)の相互作用エネルギー にパラメータ化すれば、大規模スーパーセルを用いずにSFEを評価できます [1–3]。SiC各層はSi–C二重層からなり、次の層が循環的(A→B→C)に積むか反循環的に積むかを、イジングスピン (up/down)で表します。ポリタイプの全エネルギー(1層 = 1 Si–Cペアあたり)は次式で表されます。
ここで はそれぞれ第1・第2・第3近傍の二重層間相互作用です。第3近傍まで残すと、各ポリタイプの1ペアあたりエネルギーは以下のようになります。
2H・3C・4H・6Hの4つの全エネルギーからこの連立式を解けば、 が一意に定まります [2, 4–8]。積層欠陥エネルギーは、先行部分転位の通過で生じるスピン配列の変化として評価でき、Hongらの導出 [1] により次式で与えられます。
- : 4H・6Hの固有積層欠陥エネルギー (J/m2)
- : 第4近傍ボンド間相互作用(4HのSFEを支配)
- : 第6近傍ボンド間相互作用(6HのSFEを支配)
- : (0001)面の1 Si–Cペアあたり面積、 は面内格子定数
4HのSFEは短距離の に、6HのSFEはより長距離の に比例します。一般に であるため、 となることが本手法から自然に導かれます。
計算モデルと計算条件#
計算モデルとして、共通の六方晶セルで2H・3C・4H・6Hの4ポリタイプを作成しました(図1)。すべて面内格子定数 を共通とし、二重層数に応じてc軸長を変えています。これにより逆空間のc*方向のk点密度を全ポリタイプで揃えられます(数meV/ペアの微小なエネルギー差を精密に取り出すために本質的に重要)。各構造はAdvance/PHASE同梱のPythonライブラリASE [9] を介して構造最適化(体積・c/a同時スキャン)を行いました。
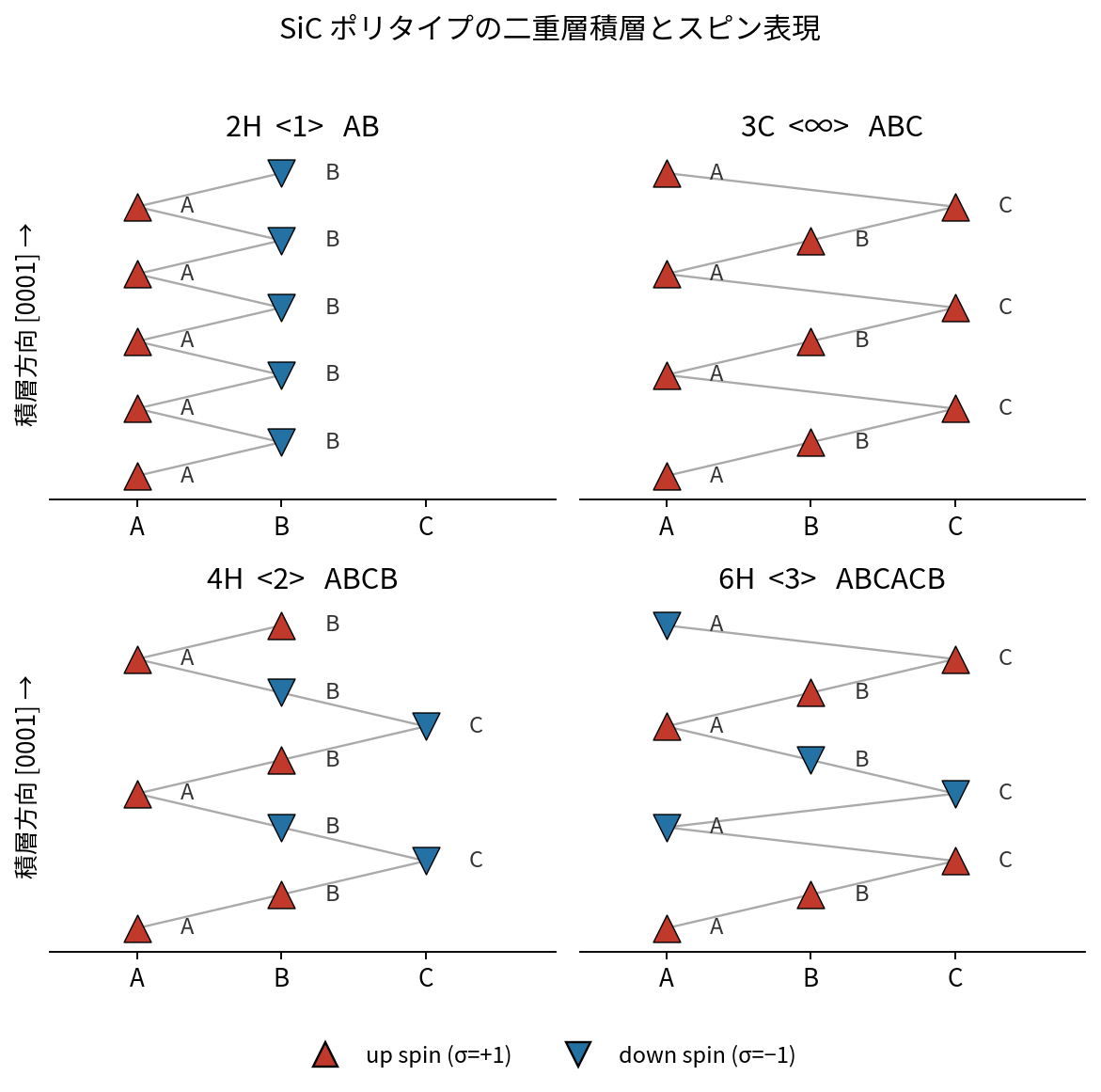
図1. SiC各ポリタイプの二重層積層とANNNIスピン表現。積層方向[0001]に沿って、面内サイトA/B/Cの並びと各層のup(▲)/down(▼)スピンを示す。2H=↑↓、3C=↑↑↑、4H=↑↑↓↓、6H=↑↑↑↓↓↓。
表1. 対象ポリタイプ
| ポリタイプ | 二重層積層 | スピン列 | Zhdanov記法 | Si–Cペア数 |
|---|---|---|---|---|
| 2H (wurtzite) | AB | ↑↓ | ⟨1⟩ | 2 |
| 3C (zincblende) | ABC | ↑↑↑ | ⟨∞⟩ | 3 |
| 4H | ABCB | ↑↑↓↓ | ⟨2⟩ | 4 |
| 6H | ABCACB | ↑↑↑↓↓↓ | ⟨3⟩ | 6 |
本解析で用いた主な計算条件を表2に示します。擬ポテンシャル・カットオフ・交換相関汎関数・面内k点を4ポリタイプで厳密に統一し、系統誤差を相殺させています。
表2. 計算条件の概要
| 項目 | 設定 |
|---|---|
| 擬ポテンシャル | Si: ノルム保存 / C: ウルトラソフト |
| 交換相関汎関数 | GGA (PBE) |
| 波動関数のカットオフエネルギー | 36 Rydberg (約490 eV) |
| 電荷密度のカットオフエネルギー | 324 Rydberg |
| k点サンプリング | 12×12×nz (nz×二重層数 = 12 で統一) |
| SCF収束条件 | 全エネルギー差 1×10−10 hartree |
| 構造最適化 | 体積・c/aスキャン + 内部座標緩和 (EOSフィット) |
計算結果と考察#
平衡構造とエネルギー#
各ポリタイプの構造最適化で得られた平衡格子定数と全エネルギーを表3に示します。得られたc/a比は実測とよく一致し(3C = 2.4495 は理想立方構造の )、体積弾性率はいずれも約211 GPa(SiCの実測 約220 GPa)と妥当です。面内格子定数 は約3.10 Åであり、GGAの特性通り実験値(3.081 Å)をわずかに過大評価していますが、4ポリタイプ間のばらつきは0.16%以内です。
表3. 平衡構造とエネルギー(PHASE計算)
| ポリタイプ | 全エネルギー [eV/セル] | a [Å] | c/a | B [GPa] |
|---|---|---|---|---|
| 2H | −525.0191 | 3.0949 | 1.6413 | 211.3 |
| 3C | −787.5479 | 3.1000 | 2.4495 | 211.2 |
| 4H | −1050.0677 | 3.0971 | 3.2741 | 211.2 |
| 6H | −1575.1028 | 3.0980 | 4.9071 | 211.1 |
1ペアあたりのエネルギーを比べると、6Hが最安定で、4Hはわずかにエネルギーが高く(+0.21 meV/ペア)、近縮退状態にあります(3C +1.17、2H +7.58 meV/ペア)。これはSiCポリタイプがANNNIモデルの多重相縮退点近傍にあるという理論描像を、本計算が独立に再現したことを意味します。
各ポリタイプの平衡構造は、体積と c/a 比を同時にスキャンして構造最適化することで決定しました。代表例として 4H-SiC の体積–エネルギー曲線(六方晶 EOS)を図2に示します。各体積で c/a 比と内部座標を最適化し、Birch–Murnaghan 状態方程式でフィットすることで、平衡体積 ・平衡エネルギー・体積弾性率 を求めています。
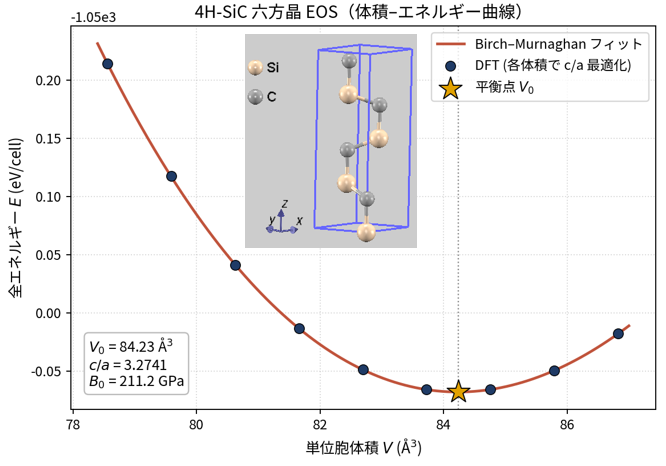
図2. 4H-SiC の六方晶 EOS(体積–エネルギー曲線)。青点は DFT 計算値(各体積で c/a 最適化済み)、実線は Birch–Murnaghan フィット、星印は平衡点 。得られた Å3、、 GPa は表3の値に対応する。
ANNNI層間相互作用パラメータ#
表3の全エネルギーから連立式を解いて得た層間相互作用を表4に示します。符号は , , と、ポリタイプ発現の条件をすべて満たしています。また は多重相縮退点 の近傍で、SiCが多数のポリタイプをもつことと整合します。
表4. ANNNI層間相互作用 (単位: meV/ペア)
| 本計算 (PBE) | 文献値 [2] (Cheng 1988, LDA) | 文献値 [4] (Limpijumnong 1998) | |
|---|---|---|---|
| +3.60 | +4.85 | +3.06 | |
| −2.08 | −2.56 | −2.57 | |
| −0.40 | −0.50 | −0.35 |
積層欠陥エネルギーの比較#
得られた からSFEを算出し、実験値および他グループの理論値と比較しました(表5、図3)。
表5. SiC積層欠陥エネルギー: 本計算と実験値の比較 (単位: mJ/m2)
| 本計算値 (PBE) | 実験値 [1] (Hong 2000) | |
|---|---|---|
| 16.1 | 14.7 ± 2.5 | |
| 3.05 | 2.9 ± 0.6 |
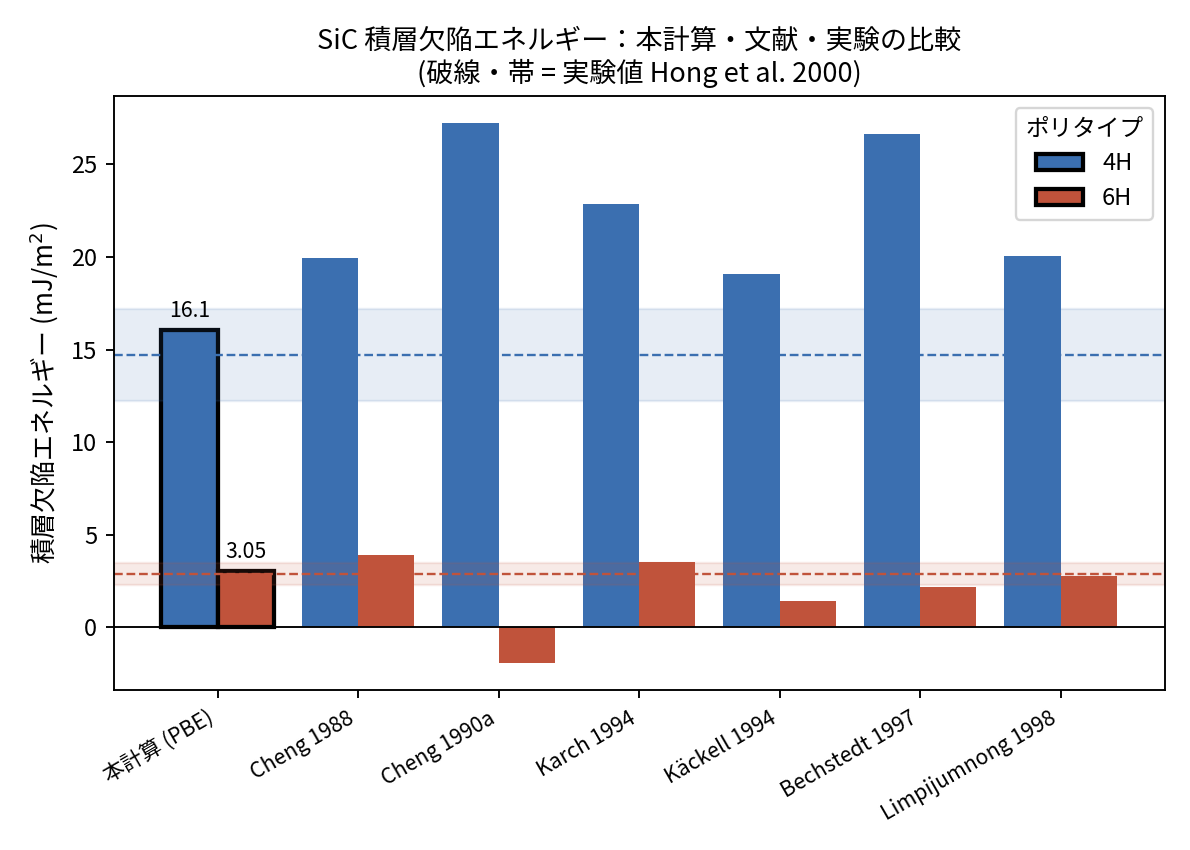
図3. SiC積層欠陥エネルギーの比較。本計算(PBE、黒枠)、既報のANNNI推定値、および実験値(破線・帯 = Hong et al. 2000)。本計算は4H・6Hとも実験の誤差帯に収まっている。各文献値の出典は参考文献 [2], [4]–[8] を参照。
本計算で得られた mJ/m2、 mJ/m2 は、いずれも実験値の誤差範囲内に収まりました。既報のANNNI推定は を19〜27 mJ/m2 と実験より大きく見積もるものが多いなか、本PBE計算は既報の中でも最も実験値に近い結果を示しています。 も実験の比(≈5.1)を再現しています。SFEに用いる格子定数 を実験値(3.081 Å)から緩和値(3.100 Å)まで変えても の変動は1%未満で、結果は頑健です。
なお、6HのSFEは微小な (約 −0.4 meV)に比例するため、 は計算精度(カットオフ・k点・SCF収束)に敏感です。本計算のエネルギー有効桁からは に ±0.3〜0.4 mJ/m2 程度の数値不確かさが見込まれますが、それでも実験帯に収まっています。3近傍近似の妥当性をより厳密に確認するには、第5のポリタイプ(例: 15R)を加えて が より十分小さいことを確認します(Cheng らは meV と報告 [2])。
補足:ANNNIモデルの適用範囲と留意点#
本手法は少数の小さなポリタイプ計算から積層欠陥エネルギーを効率的に得られる強力な近似ですが、次の前提のもとで成り立つ点に留意が必要です。適用対象を見極めることで、信頼性の高い評価が可能になります。
- 対象構造:同一の層が2通りの向き(up/downスピン)で積み重なる「ポリタイプ型」構造で、積層欠陥が結合の切断・再構成を伴わない純粋な積層変化であること(四面体配位半導体のグライド面固有欠陥、最密充填構造など)。転位芯の再構成、ダングリングボンド、荷電状態や不純物偏析を伴う欠陥は本モデルの対象外です。
- 相互作用の短距離性(打ち切り):層間相互作用 が とともに急減し、少数項(本事例では まで)で打ち切れること。SiCでは や4スピン結合が より十分小さく成立しますが [2]、振動的(Friedel型)な長距離相互作用が効く系では、15R などを追加して収束を確認する必要があります。
- 対(2スピン)相互作用が主であること:多体(4スピン)結合が無視できること。一部のZnSポリタイプのように高次項が必要な系では、モデルの拡張を要します [2]。
- 剛体層近似:各層を同一の剛体ユニットとして扱うため、局所環境に依存する層緩和や層間隔の不均一(格子緩和による相安定化機構)は近似的にしか取り込めません。
本事例のSiC(4H/6H)はこれらの前提を良く満たす典型例であり、実験と良好に一致しました。一方、金属の積層欠陥や、芯の再構成を伴う欠陥、長距離相互作用が支配的な系では、積層欠陥を含む直接スーパーセル計算など他手法との併用・相互検証が推奨されます。
まとめ#
本解析では、第一原理計算ソフトウェアAdvance/PHASEを用い、2H・3C・4H・6H SiCの全エネルギーからANNNIモデルを介して積層欠陥エネルギーを評価しました。得られた mJ/m2、 mJ/m2 は実験値(14.7 ± 2.5、2.9 ± 0.6 mJ/m2)の誤差範囲内であり、エネルギー序列(6H最安定・4H近縮退)や層間相互作用の符号・比も理論・実験と整合しました。最大でも12原子の小さなセル計算のみで、大規模スーパーセルを用いずにSiCの微小な積層欠陥エネルギーを高精度に再現できることを示しました。第一原理計算は、積層欠陥のような微小エネルギーの界面欠陥を原子レベルで定量評価し、パワー半導体をはじめとする材料開発に貢献する強力なツールです。
本解析の詳細や、研究への適用可能性に関するご相談はこちら
お問い合わせ参考文献#
- M. H. Hong, A. V. Samant, and P. Pirouz, "Stacking fault energy of 6H-SiC and 4H-SiC single crystals", Philosophical Magazine A 80, 919 (2000).
- C. Cheng, R. J. Needs, and V. Heine, "Inter-layer interactions and the origin of SiC polytypes", J. Phys. C: Solid State Phys. 21, 1049 (1988).
- P. J. H. Denteneer and W. van Haeringen, "Stacking-fault energies in semiconductors from first-principles calculations", J. Phys. C: Solid State Phys. 20, L883 (1987).
- S. Limpijumnong and W. R. L. Lambrecht, "Total energy differences between SiC polytypes revisited", Phys. Rev. B 57, 12017 (1998).
- C. Cheng, V. Heine, and R. J. Needs, "Atomic relaxation in silicon carbide polytypes", J. Phys.: Condens. Matter 2, 5115 (1990).
- K. Karch, G. Wellenhofer, P. Pavone, U. Rössler, and D. Strauch, "Structural and electronic properties of SiC polytypes", Proc. 22nd Int. Conf. on the Physics of Semiconductors (World Scientific, 1994), p. 401.
- P. Käckell, B. Wenzien, and F. Bechstedt, "Influence of atomic relaxations on the structural properties of SiC polytypes from ab initio calculations", Phys. Rev. B 50, 17037 (1994).
- F. Bechstedt, P. Käckell, A. Zywietz, K. Karch, B. Adolph, K. Tenelsen, and J. Furthmüller, "Polytypism and Properties of Silicon Carbide", Phys. Status Solidi B 202, 35 (1997).
- A. H. Larsen et al., "The atomic simulation environment—a Python library for working with atoms", J. Phys.: Condens. Matter 29, 273002 (2017).
関連ページ#
- 第一原理計算ソフトウェア Advance/PHASE
- 解析分野:ナノ・バイオ
- 産業分野:材料・化学