構造生成ツール・機械学習ポテンシャルと第一原理計算の連携による分子性結晶の構造予測#

医薬品や有機半導体に代表される分子性結晶において、特定の分子がどのようなパッキング(多形)を取るかを予測する「結晶構造予測(CSP: Crystal Structure Prediction)」は、材料設計における最重要課題の一つです。しかし、広大な探索空間を第一原理計算(DFT)のみで探索するには膨大なコストがかかります。本解析では、代表的なアミノ酸分子であるグリシンを対象とし、対称性制約下で効率的構造生成ツールPyXtalによる初期構造のサンプリング、最新の機械学習ポテンシャルMACEによる予備最適化、および第一原理計算ソフトウェアAdvance/PHASEによる高精度なエネルギー・ランドスケープ評価を組み合わせた、実用的なCSPワークフローを実証しました。
Keywords: 第一原理計算 (DFT), 機械学習ポテンシャル (MACE), 結晶構造予測 (CSP), 分子性結晶, グリシン, 多形, エネルギー・ランドスケープ
背景:CSPにおけるMI手法の導入#
従来のCSPは、古典力場による粗い探索の後にDFTで追い込む手法が一般的でしたが、古典力場では水素結合や分散力の記述精度に限界がありました。本事例では、以下のMI(マテリアルズ・インフォマティクス)ツールを採用しています。
- PyXtal [1]: 結晶構造、特に分子性結晶の構造を、対称性(空間群)の制約に基づいてランダムに生成・予測するためのPythonライブラリです。SMILES記号から対象分子を指定することが可能です。
- MACE (Message Passing Atomic Cluster Expansion) [2]: 膨大な結晶データを学習した汎用グラフニューラルネットワーク・ポテンシャルです。本ワークフローでは、商用利用に制限のあるモデル(MACE-OFF等)に代わり、産業用途での利用に適したMACE-MPを採用しました。機械学習ポテンシャル単体では微小なエネルギー差の判定に限界があるため、本手法ではMACEを高速な「事前緩和」ツールとして活用しています。
これらのツールで用意した構造リストに対して、高精度なDFT計算(Advance/PHASE)を実行し、最終的な多形間の微小なエネルギー差の判定を行います。
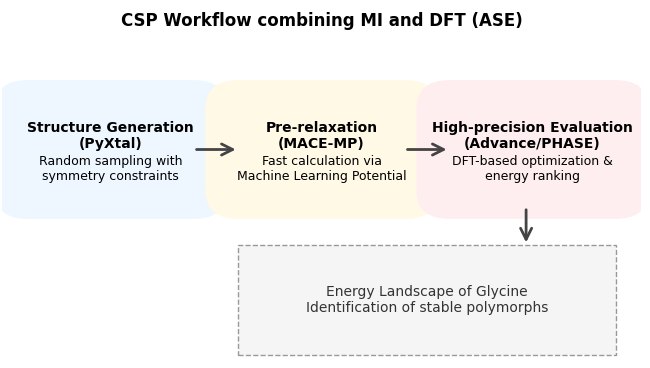
図1. 本解析におけるCSPワークフローの概念図
計算ワークフローと技術的な工夫#
1. 自動実行パイプラインの構築#
本解析では、グリシンを対象分子とします。本ワークフロー(図1)では、まず構造生成ライブラリPyXtalを用いて、グリシンで知られる主要な空間群(, , )と単位胞内の分子数を条件とし、100個の初期結晶構造をランダムに生成します。しかし、ランダム生成された直後の構造は原子同士が近すぎるなど非物理的な状態を含むことが多く、これをそのままDFT計算に投入するとSCF収束エラー等で破綻しやすくなります。
そこで、DFT計算の前処理として、汎用機械学習ポテンシャルMACEを用いた予備最適化(事前緩和)を組み込みました。PythonライブラリのASE(Atomic Simulation Environment)[3] を介してMACE (mace_mpモデル) を呼び出し、数秒〜数十秒という短時間でセル形状と原子位置をラフに最適化します。このMACEによって物理的に妥当な状態に整えられた構造を初期値として、Advance/PHASEによる高精度なフル構造最適化(ASE利用)へと自動的に引き渡すパイプラインを構築しました。このパイプラインにおいて、DFT計算をエンジンとした原子構造・セル最適化では、分子間相互作用(分散力)を正確に記述するため、PBE汎関数にDFT-D3 (BJ) 補正を組み合わせて使用しています。これにより、分子性結晶特有の緩やかなパッキングを高精度に評価することが可能となりました。
2. 極端に斜めなセルへの対応(k点メッシュの最適化)#
ランダム探索では、単位胞の角度が極端に歪んだ(例:)平坦な平行四辺形セルが生成されることがあります。従来の「セルの辺の長さ」に基づく点判定では、こうしたセルの面間隔(実際の厚み)が考慮されず、サンプリング不足による非物理的な低エネルギー状態が算出される課題がありました。
解決策:逆格子ベクトルによるk点更新パッチ
セルの実際の面間隔 (ここで は逆格子ベクトル)を算出し、これを基準に各方向の点数 を動的に決定するロジックを実装しました。
このパッチにより、歪んだセルに対しても常に物理的に適正なサンプリング密度(target_k)が保たれ、点設定不一致による数値的な誤差を排除しました。
3. ジョブ管理システムと連携したハイスループット実行制御#
ハイスループットなスクリーニングを滞りなく進めるため、計算サーバーのジョブ管理システム(PBS等)と連動するジョブ監視・制御スクリプトを構築しました。各ディレクトリの計算ステータス(実行待ち、計算中、完了、エラーなど)を常時監視し、サーバーの最大同時実行数の上限を超えないよう、キューの空き状況に応じて自動で次のDFT計算を投入します。これにより、限られた計算リソースを最大限に活用し、多数の構造最適化ジョブを自動で処理するワークフローを実現しています。
計算結果と考察#
1. エネルギー・ランドスケープ#
100種類のランダム構造から得られたエネルギー・ランドスケープ(図2)では、実験的に知られているグリシンの多形が最安定付近に正しくプロットされました。Pymatgen [4] のStructureMatcherを用いた判定により、実験構造(, 型)[5] との一致を確認しました。
なお、本事例ではワークフローの構築と動作検証を優先し、初期構造のサンプリング数を100個に制限したため、準安定相である型の抽出には至っていません。型のようなポテンシャルエネルギーの谷が狭い構造についても、実運用において初期構造の生成数をスケールアップすることで、網羅的な探索と抽出が可能となります。
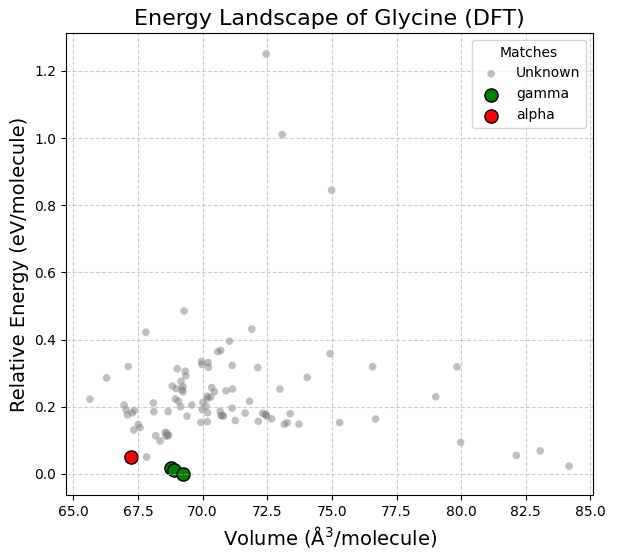
図2. グリシンのエネルギー・ランドスケープ。緑が\(\gamma\)型、赤が\(\alpha\)型を示す。DFT計算(0 K)において最も安定とされる\(\gamma\)型が、エネルギーゼロのGlobal Minimumとして正しく予測されています。
【補足:大体積・低エネルギー構造について】
図2の右下(体積 80 Å3/molecule 以上)に見られる低エネルギーのプロットは、分子が規則的な2次元シート状の水素結合ネットワークを形成する一方で、層間に大きな隙間を持ったまま局所安定化した構造です。これらは「MACEと第一原理計算が、水素結合による局所的な安定化効果を正しく記述できている証拠」と言えますが、常圧下のバルク結晶としては密度が低すぎ実在し得ないアーティファクトであるため、実際の多形評価の対象からは除外して解釈します。
2. DFTで決定した各多形の結晶構造#
Advance/PHASEを用いたDFT計算による最適化後の構造を詳細に分析すると、それぞれの多形に特有の水素結合ネットワークが原子レベルで正確に再現されていることが分かりました。視認性を高めるため、以下の構造図(図3、図4)は2×2×2のスーパーセルで表示し、その中に4つの隣接分子をハイライト(他分子は半透明化)しています。
(1) α型グリシン#
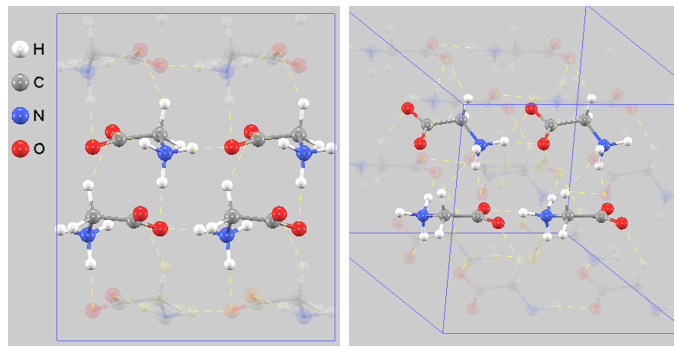
図3. \(\alpha\)型(単斜晶系 \(P2_1/c\))の結晶構造。左:正面図、右:斜視図。黄色の破線は水素結合を示す。分子がペアを組み、シート状に広がる2次元的なバイレイヤー構造が正確に再現されています。
(2) γ型グリシン#
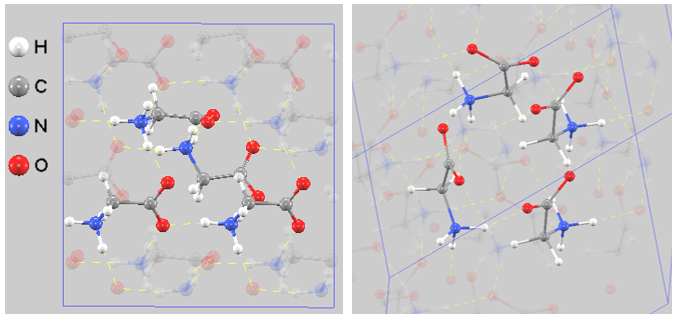
図4. \(\gamma\)型(三方晶系 \(P3_2\))の結晶構造。左:正面図、右:斜視図。分子が約120度ずつ回転しながら配置されており、3回らせん軸に沿って強固な3次元的ヘリックス状の水素結合ピラー(柱状構造)を形成している様子が確認できます。
【考察】
この「パッキングの強固さ」および「水素結合ネットワークの次元性」の違いがエネルギー差として現れています。DFT計算が、単なる格子定数の再現に留まらず、分子間の局所的な相互作用まで高度な精度で記述できていることが、これらの可視化結果からも裏付けられました。
さらに、最適化されたセル体積から分子あたりの占有体積を比較すると、型の方が型よりもわずかに小さくなります。このことから、ポテンシャルエネルギー的に最安定なのは型である一方で、結晶としての充填密度(パッキング密度)は、バイレイヤー構造を持つ型の方がわずかに高い [5] という幾何学的な特徴も確認されました。
まとめ#
本事例では、構造生成ツールPyXtal、機械学習ポテンシャルMACEとAdvance/PHASEを連携させることで、高度な専門性を要する結晶構造予測(CSP)を効率的かつ正確に実行できることを実証しました。特に、探索過程で発生する特異なセル形状に対する点メッシュの自動最適化ロジックの導入は、計算の信頼性を担保する上で有効であることが確認されました。本ワークフローは、グリシンのみならず、複雑な骨格を持つ医薬品分子や機能性有機材料の多形探索・物性予測を加速させる強力な基盤となります。
本解析の詳細や、研究への適用可能性に関するご相談はこちら
お問い合わせ参考文献#
- S. Fredericks, K. Parrish, D. Sayre, and Q. Zhu, "PyXtal: A Python library for crystal structure generation and symmetry analysis", Comp. Phys. Comm. 261, 107810 (2021).
- I. Batatia, D. P. Kovacs, G. Simm, C. Ortner, G. Csányi, "MACE: Higher order equivariant message passing neural networks for fast and accurate force fields", Advances in Neural Information Processing Systems 35, 11423 (2022).
- A. H. Larsen et al., "The Atomic Simulation Environment—A Python library for working with atoms", J. Phys.: Condens. Matter 29, 273002 (2017).
- S. P. Ong, W. D. Richards, A. Jain, G. Hautier, M. Kocher, S. Cholia, D. Gunter, V. Chevrier, K. A. Persson, and G. Ceder, "Python Materials Genomics (pymatgen) : A Robust, Open-Source Python Library for Materials Analysis", Comp. Mater. Sci. 68, 314 (2013).
- G. L. Perlovich, L. K. Hansen, and A. Bauer-Brandl, "The polymorphism of glycine. Thermochemical and structural aspects", J. Therm. Anal. Calorim. 66, 699 (2001).
関連ページ#
- 第一原理計算ソフトウェア Advance/PHASE
- 解析分野:ナノ・バイオ
- 産業分野:材料・化学