多孔質単層材料g-C3N4のCO2分子吸着特性に関する第一原理計算#

グラファイト状窒化炭素(g-C3N4)は、その低コスト、高安定性、可視光応答性から、光触媒による二酸化炭素(CO2)還元反応の材料として注目されています。CO2還元は、CO2分子が触媒表面に吸着することから始まります。この吸着過程の理解は、高性能な光触媒を設計する上で不可欠です。本解析では、第一原理計算ソフトウェアAdvance/PHASEを用い、単層g-C3N4表面へのCO2分子の吸着挙動を詳細に調査し、エネルギー的に最も安定な吸着構造と、吸着に伴う電子状態の変化を明らかにします。
Keywords: 第一原理計算, DFTシミュレーション, 吸着エネルギー, 状態密度(DOS), 部分電荷密度, 仕事関数, g-C3N4, CO2吸着, 光触媒
1. 計算モデルと計算条件#
計算モデルとして、ヘプタジンベースの単層g-C3N4を用いました(図1)。構造最適化によって得られた格子定数は7.16 Åであり、これは文献値 [1] の7.15 Åと良好に一致しています。また、真空層の厚さは文献と同様に15 Åに設定しました。
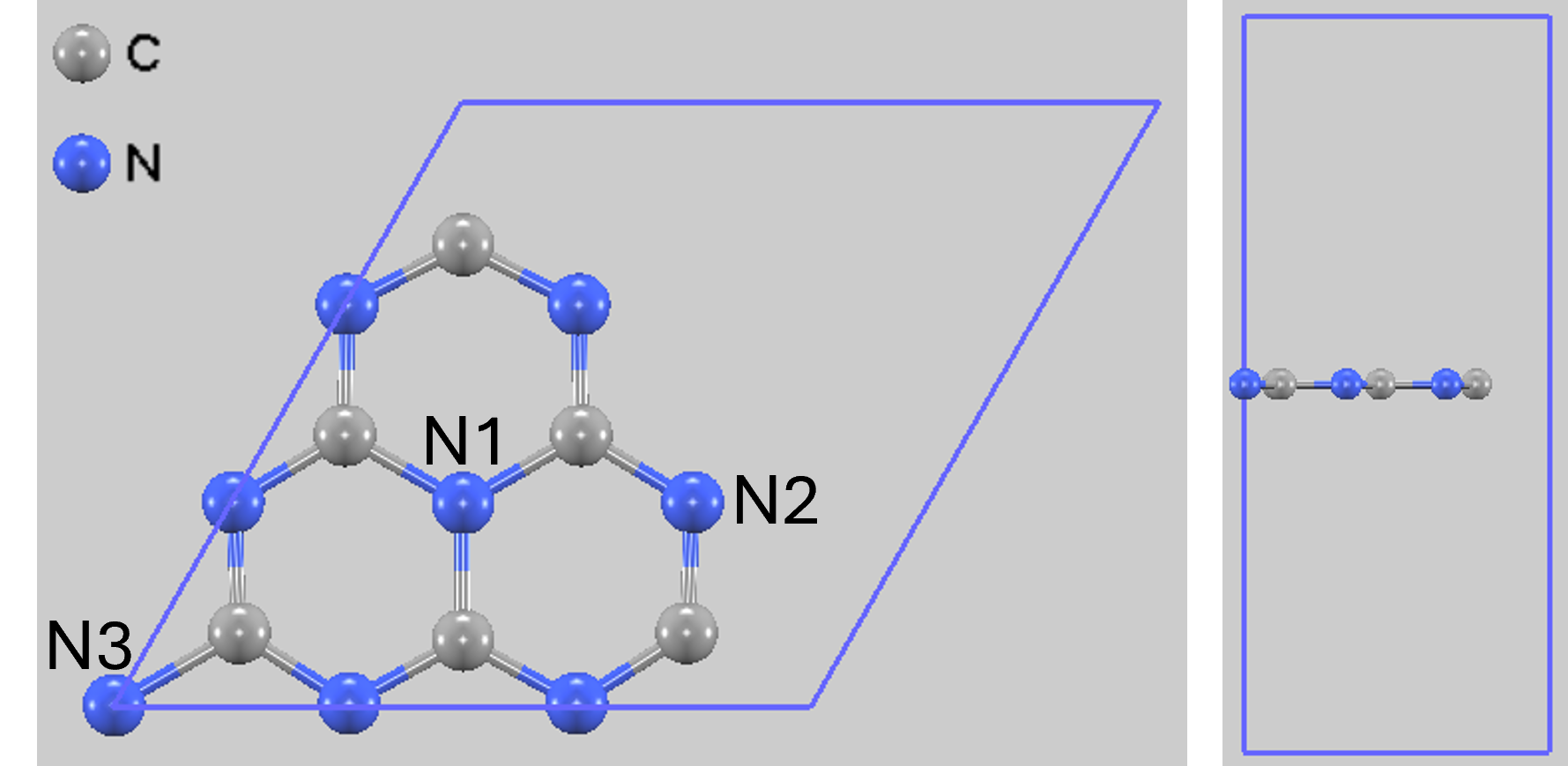
図1. 単層g-C3N4の計算モデル(左: 上面図、右: 側面図)
CO2分子の吸着サイトは、先行研究を参考に、原子上、結合上、構造の空孔部などを考慮しました。特に、構造的に異なる3種類の窒素原子サイト(図1参照)に着目しました。
- N1: ヘプタジンユニット中央に位置する3配位の窒素原子
- N2: ヘプタジンユニット外縁部に位置する2配位の窒素原子
- N3: 3つのヘプタジンユニットを連結する3配位の窒素原子
これらのサイトに対し、CO2分子を平行または垂直に配置した複数の初期構造を用意し、構造最適化計算を行いました。主な計算条件は表1の通りです。
表1. 計算条件の概要
| 項目 | 設定 |
|---|---|
| 擬ポテンシャル | ウルトラソフト擬ポテンシャル |
| 交換相関汎関数 | GGA (PBE) |
| 波動関数のカットオフエネルギー | 25 Rydberg (約340 eV) |
| k点サンプリング (SCF計算) | 4x4x1 |
計算結果と考察#
最安定吸着構造と吸着エネルギー#
様々な初期配置で構造最適化計算を行った結果、最もエネルギー的に安定な吸着構造が得られました。この構造では、ヘプタジンユニット外縁部の2配位窒素原子(N2)の近傍にCO2が吸着しています。最適化後、CO2分子はg-C3N4平面に対して傾いた配置となり、g-C3N4シート自体も平面構造から波打った(corrugated)構造へと変形しました(図2)。このような吸着による表面の再構成は、他の研究でも報告されています。
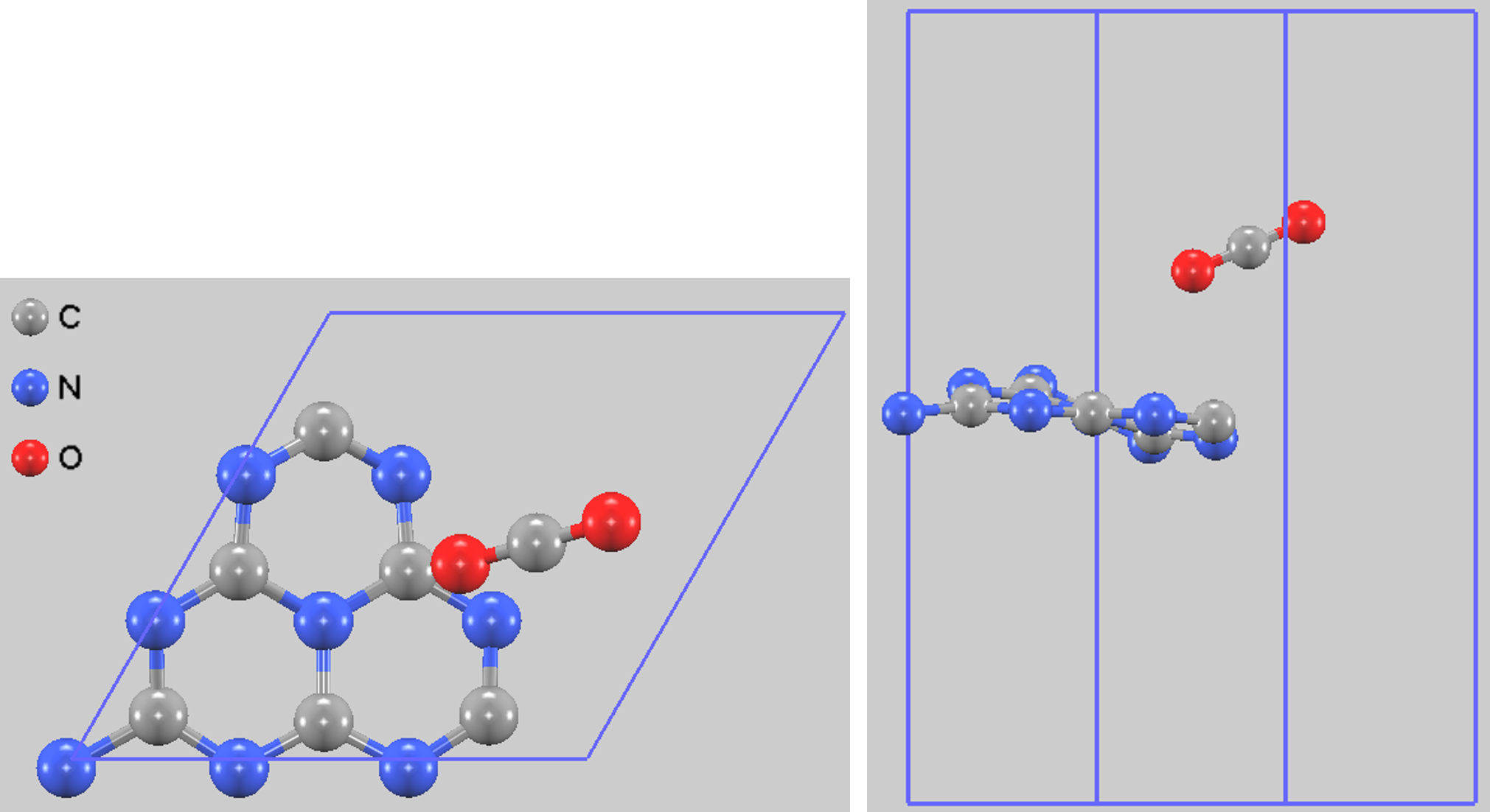
図2. 最も安定なCO2吸着構造(左: 上面図、右: 側面図)。
この最安定構造における吸着エネルギーは、以下の式で算出しました。
EAds = E(CO2-C3N4) - E(C3N4) - E(CO2)
算出された吸着エネルギーは -0.416 eV でした。負の値は、この吸着が発熱反応であり、熱力学的に安定であることを示します。この値は、参考文献[1]で報告されている-0.4181 eVと非常によく一致しており、本計算の妥当性を裏付けています。このエネルギーの大きさや吸着距離(3.2 Å)から、この吸着は物理吸着に分類されます。しかし、吸着に伴いg-C3N4シートが変形することから、比較的強い相互作用を持つ物理吸着であると考えられます。
吸着による電子状態の変化#
状態密度(DOS)とバンドギャップ#
CO2吸着がg-C3N4の電子状態に与える影響を調べるため、吸着前後の状態密度(DOS)を比較しました(図3)。吸着後、バンドギャップは1.26 eVから1.80 eVへと増大しました。これは、吸着分子と表面との相互作用により電子準位が変化したことを示唆しており、同様のバンドギャップ増大は文献でも報告されています。なお、文献 [2] によると、g-C3N4のバンドギャップ計算では実験値(約2.7 eV)をより良く再現するHSE06ハイブリッド汎関数が用いられることが多いですが、本解析で用いたGGA-PBE汎関数は、他のGGA計算結果 [3] との整合性が高く、吸着によるバンドギャップの変化の傾向も定性的に正しく捉えています。
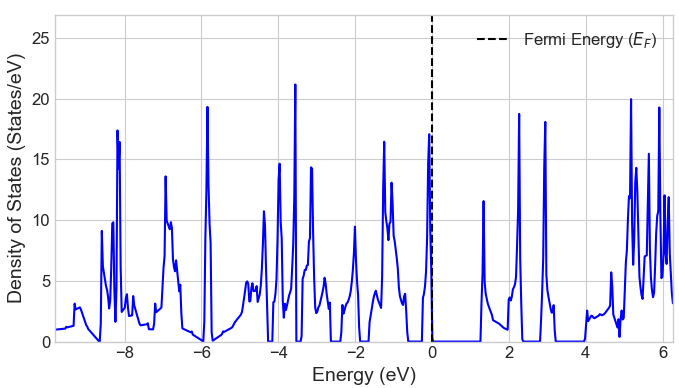
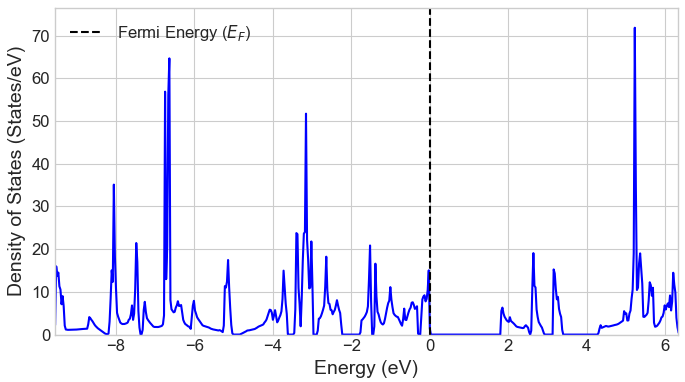
図3. CO2吸着前後の状態密度(DOS) (上: 吸着前、下: 吸着後)。フェルミエネルギー(EF)を0 eVとしています。
部分電荷密度と仕事関数#
吸着サイトの活性をより深く理解するため、価電子帯上端(占有状態)と伝導帯下端(非占有状態)の電子分布を部分電荷密度として可視化しました。吸着前のg-C3N4では、価電子帯上端(HOMO相当)は主に2配位窒素(N2)原子上に分布し、伝導帯下端(LUMO相当)は炭素原子と内部の窒素原子上に分布しています(図4)。N2原子が価電子帯の形成に大きく寄与していることが、このサイトが活性な吸着点となる一因と考えられます。
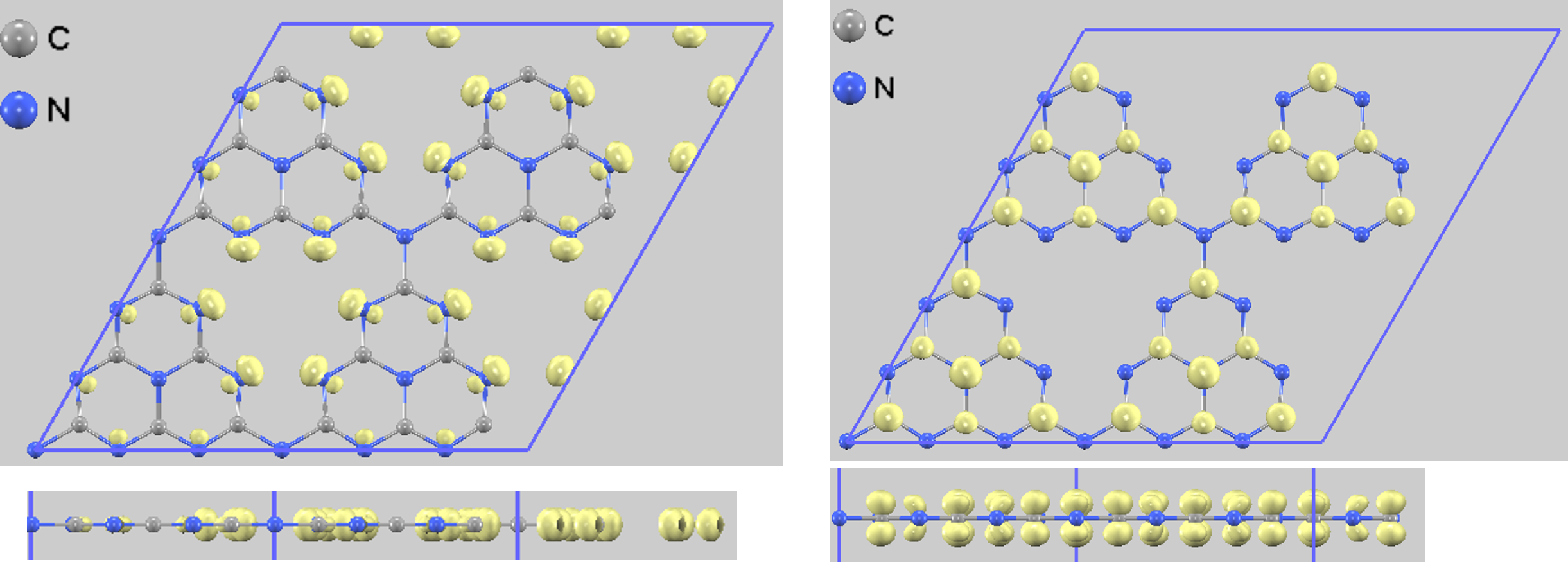
図4. 吸着前(pristine)のg-C3N4の部分電荷密度(左: HOMO側、右: LUMO側)。それぞれの電子分布を上面図と側面図で示します。
CO2吸着後は、価電子帯・伝導帯ともに電荷分布がg-C3N4側に局在しており、CO2分子自体はHOMOやLUMOの形成に直接は関与していません(図5)。これは、光励起された電子がg-C3N4の伝導帯に上がり、その後で吸着したCO2分子に移動するという、光触媒反応のメカニズムを示唆しています [4, 5]。
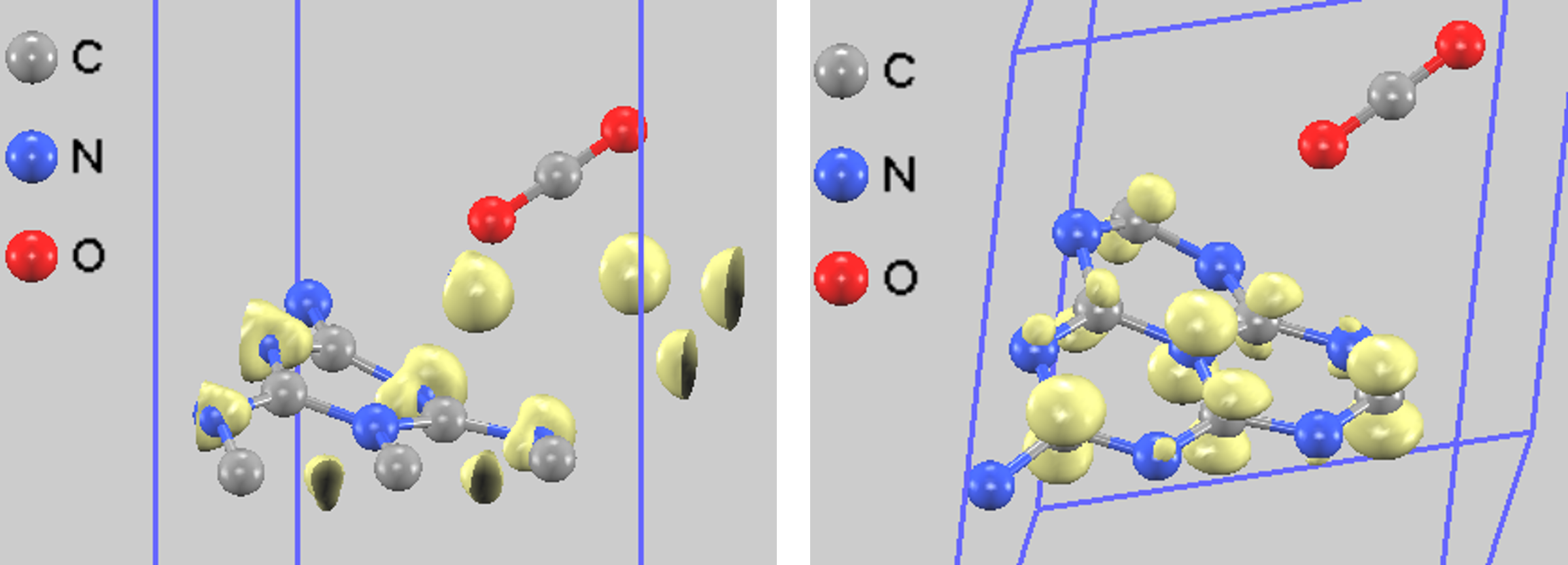
図5. CO2吸着後の部分電荷密度(左: HOMO側、右: LUMO側)。
また、電子を材料表面から真空準位まで取り出すのに必要なエネルギーである仕事関数を算出したところ、吸着前の 4.48 eV から吸着後には 5.31 eV へと増加しました(図6)。吸着による仕事関数の増加も、文献[1]の結果(4.65 eV → 5.51 eV) と定性的に一致しています。
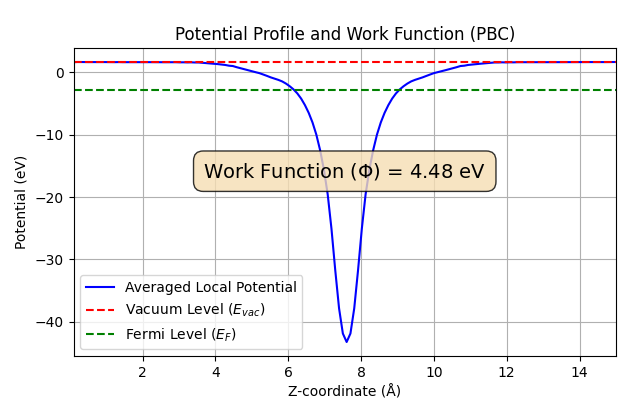
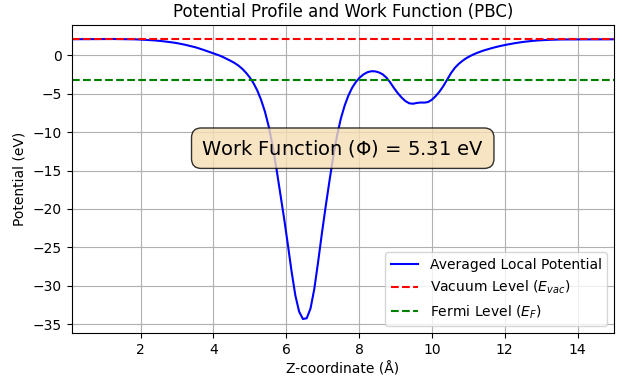
図6. CO2吸着前後の仕事関数(上: 吸着前、下: 吸着後)
まとめ#
本解析では、第一原理計算ソフトウェアAdvance/PHASEを用い、単層g-C3N4表面へのCO2分子の吸着挙動をシミュレーションしました。最もエネルギー的に安定な吸着サイトは2配位窒素原子近傍であり、吸着エネルギーは-0.416 eVと算出され、これは物理吸着を示唆する文献値とよく一致しています。最適化された構造では、g-C3N4シートが波打ち、CO2分子が傾いて吸着することが分かりました。電子状態解析の結果、CO2吸着によってバンドギャップと仕事関数が増加することが明らかになりました。また、部分電荷密度の解析から、吸着サイトであるN2原子が価電子帯上端の形成に重要であることが分かりました。さらに、吸着したCO2分子は材料のフロンティア軌道に直接寄与しないことも示唆され、これは光触媒反応における電荷移動メカニズムに関する重要な知見です。第一原理計算は、触媒設計の指針となる原子・電子レベルでの詳細な情報を得る強力なツールとなります。
本解析の詳細や、研究への適用可能性に関するご相談はこちら
お問い合わせ参考文献#
- B. Zhu, L. Zhang, D. Xu, B. Cheng, and J. Yu, "Adsorption investigation of CO2 on g-C3N4 surface by DFT calculation", Journal of CO2 Utilization 21, 327 (2017).
- B. Zhu, B. Cheng, L. Zhang, and J. Yu, "Review on DFT calculation of s-triazine-based carbon nitride", Carbon Energy 1, 32 (2019).
- J. Cui, S. Liang, X. Wang, and J. Zhang, "First principle modeling of oxygen-doped monolayer graphitic carbon nitride", Materials Chemistry and Physics 161, 194 (2015).
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong, and S. P. Chai, "Graphitic carbon nitride (g-C3N4)-based photocatalysts for artificial photosynthesis and environmental remediation: are we a step closer to achieving sustainability?", Chemical Reviews 116, 7159 (2016).
- J. Fu, B. Zhu, C. Jiang, B. Cheng, W. You, and J. Yu, "Hierarchical porous O-doped g-C3N4 with enhanced photocatalytic CO2 reduction activity", Small 13, 1603938 (2017).
関連ページ#
- 第一原理計算ソフトウェア Advance/PHASE
- 解析分野:ナノ・バイオ
- 産業分野:材料・化学