ナノシート(2D)の剥離エネルギーの第一原理計算:遷移金属ダイカルコゲナイドの例#

グラフェンに代表される原子層厚さのナノシート(2D 材料)は、層状結晶を1層まで剥がす「剥離 (exfoliation)」によって得られます。1層を剥離するのに要するエネルギー(剥離エネルギー)は、その材料が実際にどれだけ容易に単層化できるかを示す基本的な指標であり、新しい2D材料を探索するうえで重要な物理量です。層間はファンデルワールス (van der Waals, vdW) 力で弱く結合しているため、剥離エネルギーを精度良く求めるには、vdW相互作用を取り込んだ第一原理計算が必要になります。本解析では、第一原理計算ソフトウェア Advance/PHASE を用いて、代表的な遷移金属ダイカルコゲナイド (TMDC) 4種(半導体3種+金属1種)の剥離エネルギーを計算し、公開データベース JARVIS-DFT および高精度な RPA 計算の値と比較します。
Keywords: 第一原理計算, DFTシミュレーション, 2D材料, ナノシート, 剥離エネルギー, ファンデルワールス, 遷移金属ダイカルコゲナイド, マテリアルズインフォマティクス
剥離エネルギーの定義と計算方法#
剥離エネルギー は、層状結晶(バルク)から1層(単層)を取り出す際の、原子1個あたりのエネルギー変化として定義されます。JARVIS-DFT では、バルクと単層の全エネルギーをそれぞれ原子数で規格化し、その差として算出しています [1]。
- , : 単層モデルの全エネルギーと単位胞内の原子数
- , : バルク(層状結晶)モデルの全エネルギーと単位胞内の原子数
単層は層間結合を失った分だけバルクよりエネルギーが高いため、 は正の値になります。値は meV/atom で表します。JARVIS-DFT では、剥離エネルギーが 200 meV/atom 未満の材料を「容易に剥離可能」の目安としており、多くの層状材料は 60〜100 meV/atom の範囲に分布します [2]。
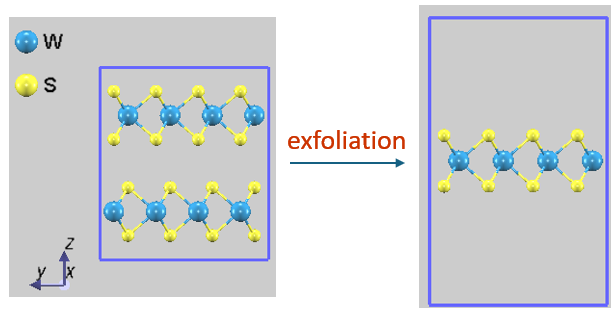
図1. WS2 を例とした剥離のモデル。(左) バルク結晶、(右) 1層のみを残し真空層を設けた単層モデル。視認性のため、それぞれ4x4x1スーパーセルを表示しています。
層間のvdW引力は標準的なGGA汎関数では十分に記述できないため、剥離エネルギーの評価にはvdW補正が不可欠です。JARVIS-DFT は非局所vdW汎関数 vdW-DF-OptB88(OptB88vdW)[3] を用いています。本解析では、GGA (PBE) に DFT-D3 分散力補正 [4] を加え、層間のvdW結合を取り込みます。
計算手順#
剥離エネルギーは、バルクと単層をそれぞれ同一条件で計算して求めます。
- バルク:Materials Project [5] から当該材料ID(表1のMP ID。例:WS2 は mp-224, バルク P63/mmc)の構造を取得し、DFT+D3 でセル/構造最適化・全エネルギー計算を行う。
- 単層:バルク構造から1層のみを残し、面直方向(c軸方向)に 15 Å 以上の真空層を設けて、同じく DFT+D3 で構造最適化・全エネルギー計算を行う。
- 両モデルの原子1個あたりの全エネルギーの差から、上式により剥離エネルギー を求める。
計算対象の選定:Materials minerによる2D材料マイニング#
計算対象は、Advance/PHASE の GUI 環境である PHASE Viewer のプラグイン「Materials miner」の 2D materials 機能を用いて選定しました。この機能は、NISTが公開する JARVIS-DFT の2D材料データセットを取得し、剥離エネルギー・バンドギャップ・生成エネルギーなどの物性を散布図として可視化します(図2)。
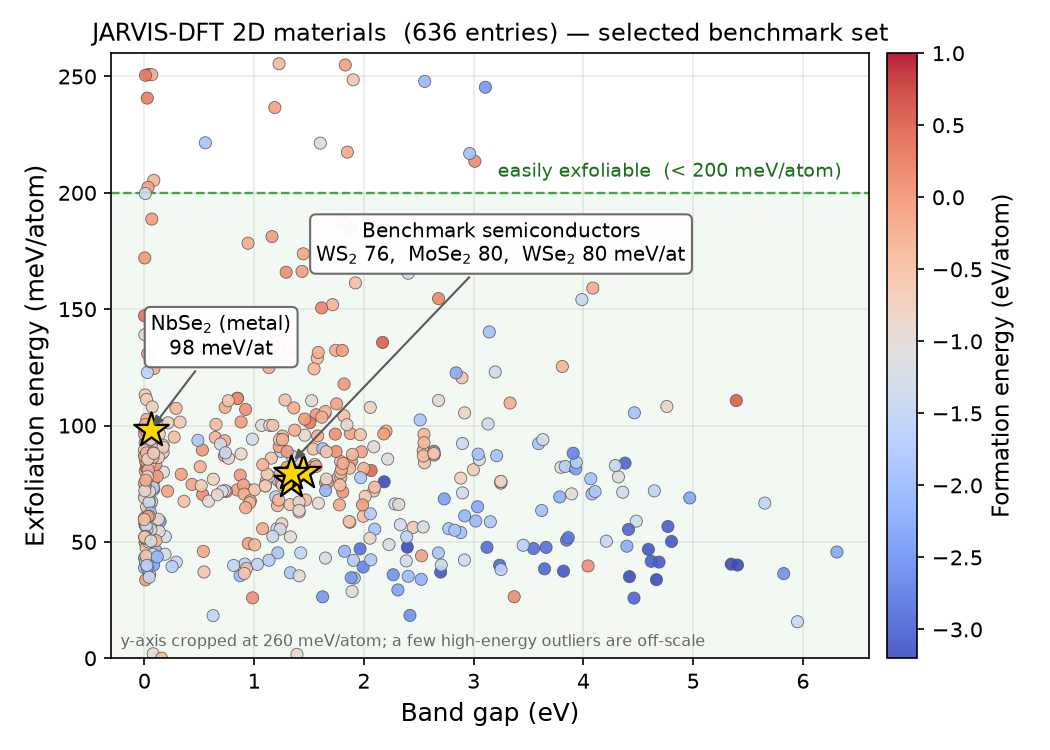
図2. Materials miner (2D materials) で可視化した JARVIS-DFT の2D材料。横軸はバンドギャップ、縦軸は剥離エネルギー(色は生成エネルギー)。本解析で選定した4材料(★, 半導体3種+金属1種)を示します。いずれも「容易に剥離可能」(< 200 meV/atom)の領域にあります。縦軸は見やすさのため260 meV/atomで切っており、少数の高エネルギー外れ値は範囲外です。
この分布から、以下の4種の遷移金属ダイカルコゲナイド(半導体3種+金属1種)を選びました。剥離エネルギーはいずれも 76〜98 meV/atom と、典型的なvdW層状材料の範囲にあります。
表1. 選定した4材料と JARVIS-DFT 参照データ
| 材料 | JARVIS ID / MP ID | バンドギャップ (eV) | 電子物性 |
|---|---|---|---|
| WS2 | JVASP-658 / mp-224 | 1.34 | 半導体 |
| MoSe2 | JVASP-649 / mp-1634 | 1.45 | 半導体 |
| WSe2 | JVASP-652 / mp-1821 | 1.33 | 半導体 |
| NbSe2 | JVASP-655 / mp-2207 | 0.06 | 金属 |
※NbSe2のバンドギャップはJARVIS-DFTのデータ上0.06 eVとなっていますが、実質的には金属(ギャップゼロ)です。
Materials miner で候補材料を絞り込んだのち、対応する Materials Project ID(表1)から結晶構造を取得します。このバルク構造と、そこから作成した単層モデルをそれぞれ Advance/PHASE(DFT+D3)で計算し、剥離エネルギーを算出します。
計算条件#
バルクと単層は同一の計算条件で計算し、系統誤差を打ち消します。本解析で用いた主な計算条件を表2に示します。単層モデルには、層間相互作用を排除するため面直方向に15 Å以上の真空層を設けます。なお、バルクの構造最適化には六方晶EOSを利用して格子定数を最適化します。
表2. 計算条件の概要
| 項目 | 設定 |
|---|---|
| 擬ポテンシャル | ウルトラソフト擬ポテンシャル |
| 交換相関汎関数 | GGA (PBE) |
| ファンデルワールス補正 | DFT-D3 |
| 波動関数のカットオフエネルギー | 40 Rydberg |
| k点サンプリング(例) | 10×10×2(バルク)、10×10×1(単層) |
NbSe2 は金属であり、フェルミ面付近の電子状態を正確に記述するために、k点数やバンド数を十分に大きく設定します。金属層状材料では層間のvdW結合に加えて金属結合的な寄与も加わりますが、剥離エネルギーはあくまで層間の結合の強さを反映します。
計算結果と考察#
PBE+D3で計算した各材料の剥離エネルギーを、JARVIS-DFT の参照値および高精度な RPA 計算値 [6] と並べて表3・図3に示します。
表3. 剥離エネルギーの比較(単位: meV/atom)
| 材料 | JARVIS-DFT (OptB88vdW) [2] | RPA [6] (高精度) | Advance/PHASE (PBE+D3, 本計算) |
|---|---|---|---|
| WS2 | 76.3 | 58.5 | 54.0 |
| MoSe2 | 80.2 | 64.0 | 66.7 |
| WSe2 | 79.6 | 62.7 | 71.8 |
| NbSe2 | 98.3 | — | 71.1 |
※ RPA値は文献[6](group-6 TMDC二層)を単層・原子あたりに換算(×2)した値。NbSe2 は文献値なし。
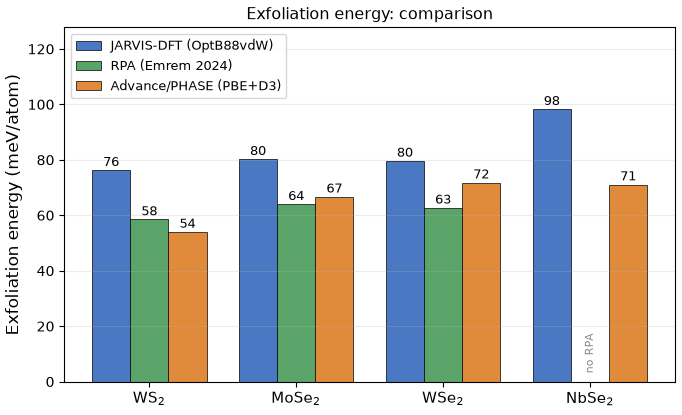
図3. 選定4材料の剥離エネルギー。JARVIS-DFT(青, OptB88vdW)、RPA(緑, 高精度ベンチマーク [6])、Advance/PHASE(橙, PBE+D3)の比較。半導体3種(WS2, MoSe2, WSe2)ではPBE+D3はRPAとよく一致します(金属のNbSe2は文献RPA値なし)。
4材料の結果:Advance/PHASE(PBE+DFT-D3)で得られた剥離エネルギーは、WS2 54.0、MoSe2 66.7、WSe2 71.8、NbSe2 71.1 meV/atom でした。いずれも JARVIS-DFT(OptB88vdW)より約10〜29% 低いものの、全材料が「容易に剥離可能」の目安(< 200 meV/atom)を大きく下回る数十 meV/atom のオーダーにあり、vdW結合した層状物質であるという描像は共通です。構造面でも、PBE+D3での単層面内格子定数(例 WS2 a = 3.21 Å, MoSe2 3.32 Å, NbSe2 3.47 Å)は JARVIS 値と各材料でよく一致します。
高精度RPAとの比較: RPA(乱雑位相近似)を用いた高精度計算 [6] を参照しました。文献[6]は二層あたりの値なので、同一基準(単層・原子あたり)に直すと、RPA の剥離エネルギーは WS2 58.5、MoSe2 64.0、WSe2 62.7 meV/atom です。PBE+D3での結果(54.0 / 66.7 / 71.8)はこの高精度RPAに ±15% 以内(平均約9%)で一致しています。
層状物質の剥離エネルギーは用いるvdW手法に敏感で、手法間で10〜20 meV/atom 程度ばらつくことが知られています。面積あたりで見ると PBE+D3 は 0.29〜0.36 J/m2 で、TMDCへき開エネルギーの文献値(約0.27〜0.42 J/m2)の範囲に収まります。
まとめ#
本解析では、第一原理計算ソフトウェア Advance/PHASE を用いて、JARVIS-DFT の2D材料データに含まれる遷移金属ダイカルコゲナイド4種(半導体 WS2, MoSe2, WSe2 +金属 NbSe2)の剥離エネルギーを PBE+DFT-D3で計算しました。その結果(54〜72 meV/atom)は、JARVIS-DFT(OptB88vdW)と同じスケールであり、高精度RPA計算ともよく一致しています。第一原理計算とマテリアルズインフォマティクスを組み合わせることで、2D材料の探索から高精度評価までのプロセスを大幅に効率化し、次世代デバイス向けの新規材料開発を加速することが期待されます。
本解析の詳細や、研究への適用可能性に関するご相談はこちら
お問い合わせ参考文献#
- K. Choudhary, I. Kalish, R. Beams, and F. Tavazza, "High-throughput Identification and Characterization of Two-dimensional Materials using Density functional theory", Sci. Rep. 7, 5179 (2017).
- K. Choudhary et al., "The joint automated repository for various integrated simulations (JARVIS) for data-driven materials design", npj Comput. Mater. 6, 173 (2020). データベース: https://jarvis.nist.gov/
- J. Klimeš, D. R. Bowler, and A. Michaelides, "Chemical accuracy for the van der Waals density functional", J. Phys.: Condens. Matter 22, 022201 (2010).
- S. Grimme, J. Antony, S. Ehrlich, and H. Krieg, "A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu", J. Chem. Phys. 132, 154104 (2010).
- A. Jain et al., "Commentary: The Materials Project: A materials genome approach to accelerating materials innovation", APL Mater. 1, 011002 (2013). データベース: https://materialsproject.org/
- B. Emrem, J.-O. Joswig, and T. Heine, "Precise structure and energy of group 6 transition metal dichalcogenide homo- and heterobilayers in high-symmetry configurations", 2D Mater. 11, 035011 (2024).
関連ページ#
- 第一原理計算ソフトウェア Advance/PHASE
- 解析分野:ナノ・バイオ
- 産業分野:材料・化学