間接-直接バンドギャップ遷移:MoS2の単層化による電子構造の変化の解析#

グラフェンに代表される二次元物質は、その特異な物理的・化学的特性から次世代の電子・光学デバイス材料として精力的に研究されています。中でも、遷移金属ダイカルコゲナイドの一種である二硫化モリブデン(MoS2)は、層数によってその電子的性質が劇的に変化することで知られています。バルク状態では間接遷移型の半導体ですが、単原子層(モノレイヤー)にまで薄くすると直接遷移型の半導体へと変化し、発光効率が著しく向上します。この現象は、MoS2をLEDや高効率トランジスタといった多様な光・電子デバイスへ応用する上で極めて重要です。本解析では、第一原理計算ソフトウェアAdvance/PHASEを用いて、MoS2のバルクおよび単層モデルのバンド構造を計算し、この層数制御による電子構造変化のメカニズムを明らかにします。
Keywords: 第一原理計算, DFTシミュレーション, 二硫化モリブデン(MoS2), バンド構造, 二次元物質, 直接・間接遷移
計算モデル#
計算対象として、バルク状態のMoS2(空間群: P6₃/mmc)と、そこから一層だけを抜き出して作成した単層MoS2モデルの2種類を用意しました。バルクおよび単層構造のモデルを図1に示します。単層モデルでは、周期境界条件の下で隣接する層との相互作用を排除するため、c軸方向(層の垂直方向)に十分な真空層(15 Å)を設けています。
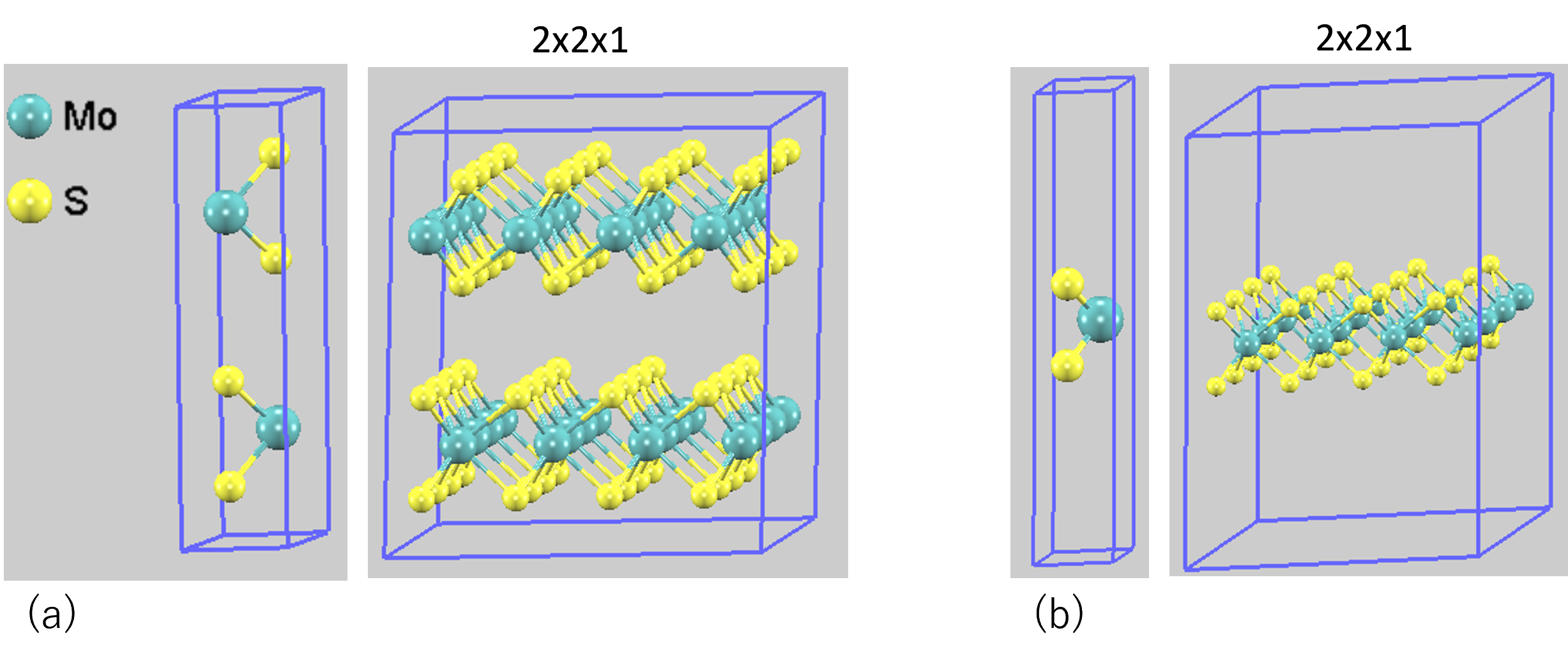
図1. MoS2の計算モデル: (a)バルク (b)単層。計算にはprimitive cellを使用し、図では構造を視覚的に分かりやすくするため2x2x1スーパーセルも示しています。
主な計算条件は表1に示します。
表1. 主な計算条件
| 項目 | 設定値 |
|---|---|
| 擬ポテンシャル | ウルトラソフト擬ポテンシャル |
| 交換相関汎関数 | GGA-PBE |
| 波動関数カットオフ | 25 Rydberg |
| k点メッシュ | バルク: 9x9x5 単層: 9x9x1 |
計算結果と考察#
電子状態密度:バルクと単層の比較#
図2と図3に、それぞれバルクと単層MoS2の電子状態密度(DOS)を示します。両者を比較すると、バルクから単層になることで、フェルミ準位(エネルギー0 eV)近傍の禁制帯(バンドギャップ)が明確に増大していることが確認できます。
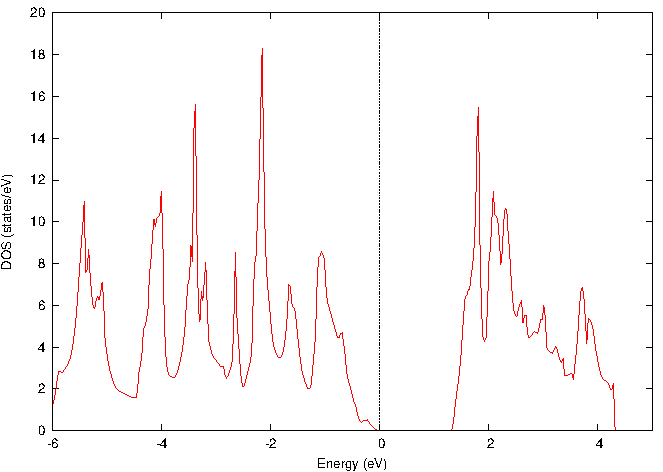
図2. バルクMoS2の電子状態密度(DOS)
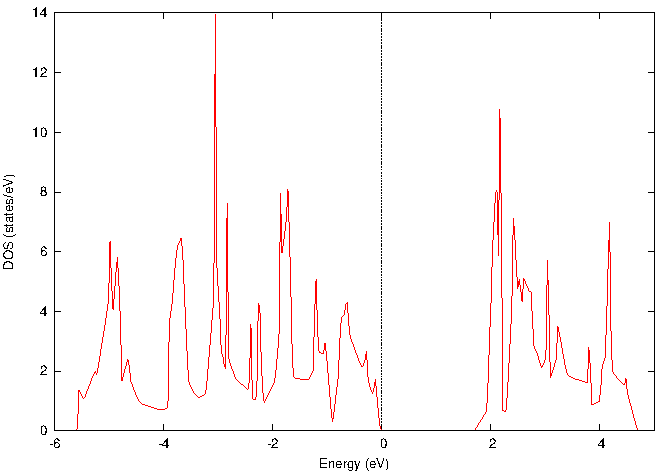
図3. 単層MoS2の電子状態密度(DOS)
電子バンド構造:バルクと単層の比較#
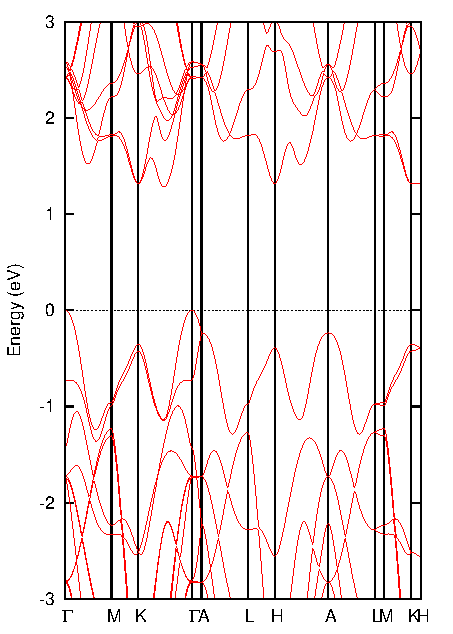
図4. バルクMoS2のバンド構造
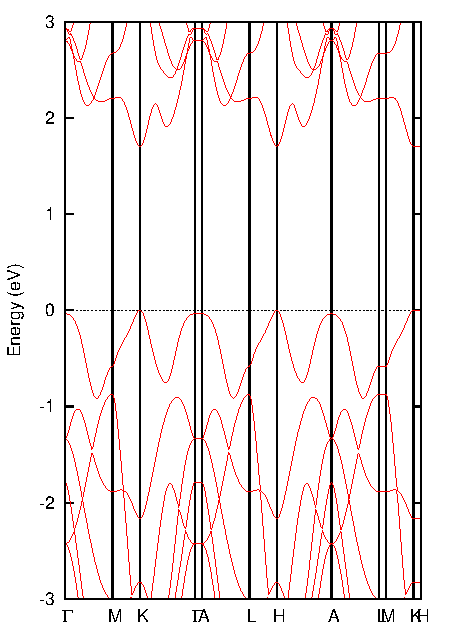
図5. 単層MoS2のバンド構造
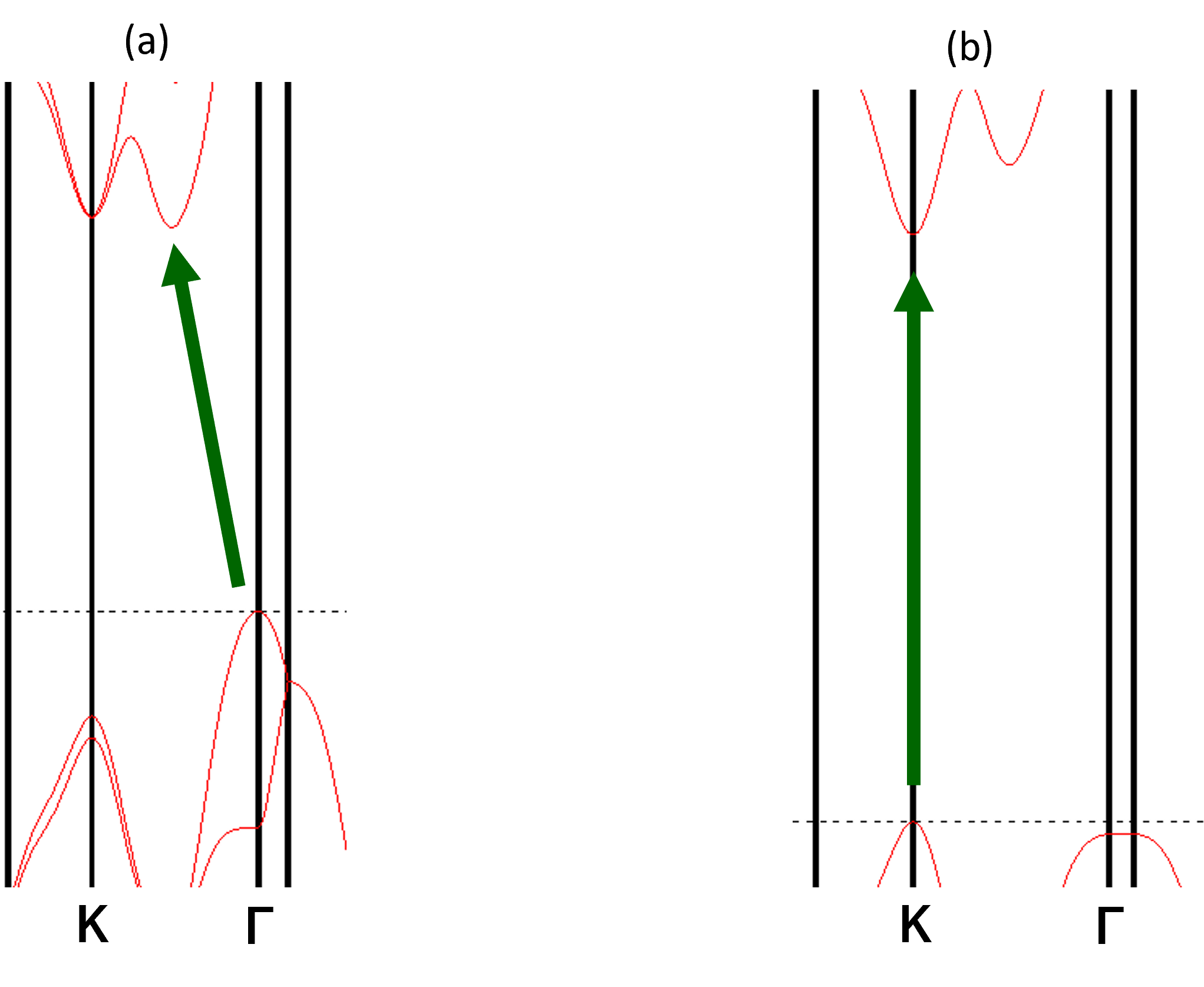
図6. バンドギャップ近傍の拡大図。 (a) バルクでは、価電子帯の頂上(VBM, Γ点)と伝導帯の底(CBM, Γ-K間)の位置が異なり、間接遷移となります。 (b) 単層では、VBMとCBMが共にK点に位置し、直接遷移となります。
層数の変化が電子構造に与える影響を理解するために、バルクと単層MoS2のバンド構造(図4、図5)を比較します。
-
バルクMoS2: 図4と図6(a)から、価電子帯の頂上(VBM)はΓ点に位置する一方、伝導帯の底(CBM)はΓ点とK点を結ぶ経路上に存在することが分かります。VBMとCBMがブリルアンゾーン内の異なる波数ベクトル(k点)にあるため、バルクMoS2は間接遷移半導体となります。バンドギャップは1.297 eVと計算されました。
-
単層MoS2: 図5と図6(b)から、VBMとCBMが共にブリルアンゾーンのK点に位置していることが明瞭に見て取れます。これにより、単層MoS2は直接遷移半導体へと変化します。バンドギャップも1.680 eVに増大しています。この直接遷移型への変化が、単層MoS2の高い発光効率の起源となります。
文献値との比較: 本計算で得られたバンド構造の定性的特徴(間接→直接ギャップ遷移)および定量的特徴(バルクで約1.30 eV、単層で約1.68 eVのギャップ値)は、実験報告や他の理論計算と良く一致しています [1, 2]。これにより、本シミュレーションがMoS2の基本的な電子物性を正しく再現していることが検証されました。
ここで単層モデルのバンド構造(図5)に注目すると、K点とH点におけるバンドのエネルギー準位がほぼ縮退していることがわかります。ブリルアンゾーンにおいて、K点とH点は面内方向の座標 (, ) が同じで、面直方向の座標 () のみが異なる高対称点です。単層モデルでは、c軸方向に大きな真空層を設けることで層間の電子的な相互作用を遮断しているため、電子バンドは面直方向(バンド図上のK-H経路)の波数にほとんど依存しなくなります。その結果、バンドの分散がフラットになり、シミュレーションが物質の2次元的な性質を正しく捉えられていることが示唆されます。
まとめ#
本解析では、第一原理計算ソフトウェアAdvance/PHASEを用い、二硫化モリブデン(MoS2)の電子的性質が層数によって劇的に変化するメカニズムを明らかにしました。計算結果は、バルク状態の間接ギャップ半導体から単層状態の直接ギャップ半導体への遷移を明確に示し、実験的に知られる物性変化を再現することに成功しました。このように、第一原理計算は、ナノ材料が示す複雑で興味深い物理現象を原子・電子レベルから解明し、新しい機能性材料の設計やデバイス開発に不可欠な指針を与える強力なツールです。
本解析の詳細や、研究への適用可能性に関するご相談はこちら
お問い合わせ参考文献#
-
A. Splendiani, L. Sun, Y. Zhang, T. Li, J. Kim, C. Y. Chim, G. Galli, and F. Wang, "Emerging photoluminescence in monolayer MoS2", Nano lett. 10, 1271 (2010).
-
K. F. Mak, C. Lee, J. Hone, J. Shan, and T. F. Heinz, "Atomically Thin MoS2: A New Direct-Gap Semiconductor", Phys. Rev. Lett. 105, 136805 (2010).
関連ページ#
- 第一原理計算ソフトウェア Advance/PHASE
- 解析分野:ナノ・バイオ
- 産業分野:材料・化学