MOFへの水素吸蔵シミュレーション#

水素社会の実現に向け、水素を分子のまま吸蔵・輸送する手段としてMetal-Organic Framework (MOF)が検討されています。この特性を持つ物質を探索する上で、MOF内で水素分子がどのように安定的に吸蔵されるかを理解することが不可欠です。この点を明らかにするため、第一原理計算ソフトウェアAdvance/PHASEを用いてMOFの一種であるNi2(m-dobdc)を対象とし、水素分子の吸蔵状態のシミュレーションを通じて活性点や電子状態の解析を行いました。
Keywords: First-principles calculation (DFT), Metal-Organic Framework, hydrogen storage
計算モデル・計算条件#
第一原理計算によるNi2(m-dobdc)への水素分子吸着のエネルギーを調べるために、初めにMOFの一種であるNi2(m-dobdc)の安定構造をシミュレーションします。論文 [1] に報告されている構造を初期構造とします。その格子定数は、a = b = 2.5797 nm, c = 0.6792 nm, α = β = 90°, γ = 120°です。また、シミュレーションに用いた主な計算条件は表1の通りです。
表1.シミュレーションに用いた計算条件
| 平面波基底のカットオフエネルギー [Hartree] | 波動関数 | 12.5 |
|---|---|---|
| 電荷密度 | 112.5 | |
| サンプリングk点 | 2×2×4 一様メッシュ | |
| 収束条件 | SCF | 1.0×10-8 [Hartree/atom] |
| 原子位置の最適化 | 1.0×10-3 [Hartree/Bohr] | |
| 交換・相関エネルギー汎関数 | 一般化勾配近似 PBE汎関数 | |
| van der Waals補正 | DFT-D3 | |
Ni2(m-dobdc)の安定構造#
図1に構造最適化の結果を示します。この構造のオープンメタルサイト(Ni原子のサイト)に水素分子を吸着させ、水素分子の吸蔵エネルギーを求めることになります。閉殻の電子構造を持つ分子が吸着する場合、化学的な結合はできにくく、van der Waals力のような弱い力で吸着していることが考えられるので、表1にあるようにDFT-D3によるvan der Waals補正 [2] を考慮したシミュレーションを行っています。
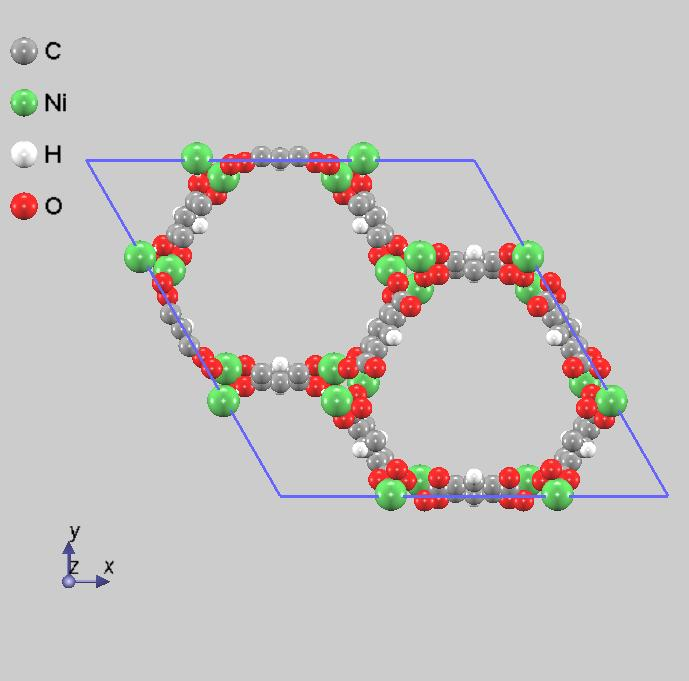
図1.最適化したNi2(m-dobdc)の構造
水素分子を吸蔵したNi2(m-dobdc)の構造最適化#
まず、1個のオープンメタルサイトに水素分子が吸着している場合の構造最適化を行いました。図2に結果を示します。赤丸で囲った分子が吸蔵されている水素分子です。また、吸蔵された水素分子の水素原子間距離や水素原子-金属原子間距離を表2に示します。孤立した水素分子の水素原子間距離は、0.075 [nm]であるので、結果はわずかに長くなっていますが、有意な差であるとは考えにくい量であり、分子のままで吸蔵されている結果となっていると考えられます。
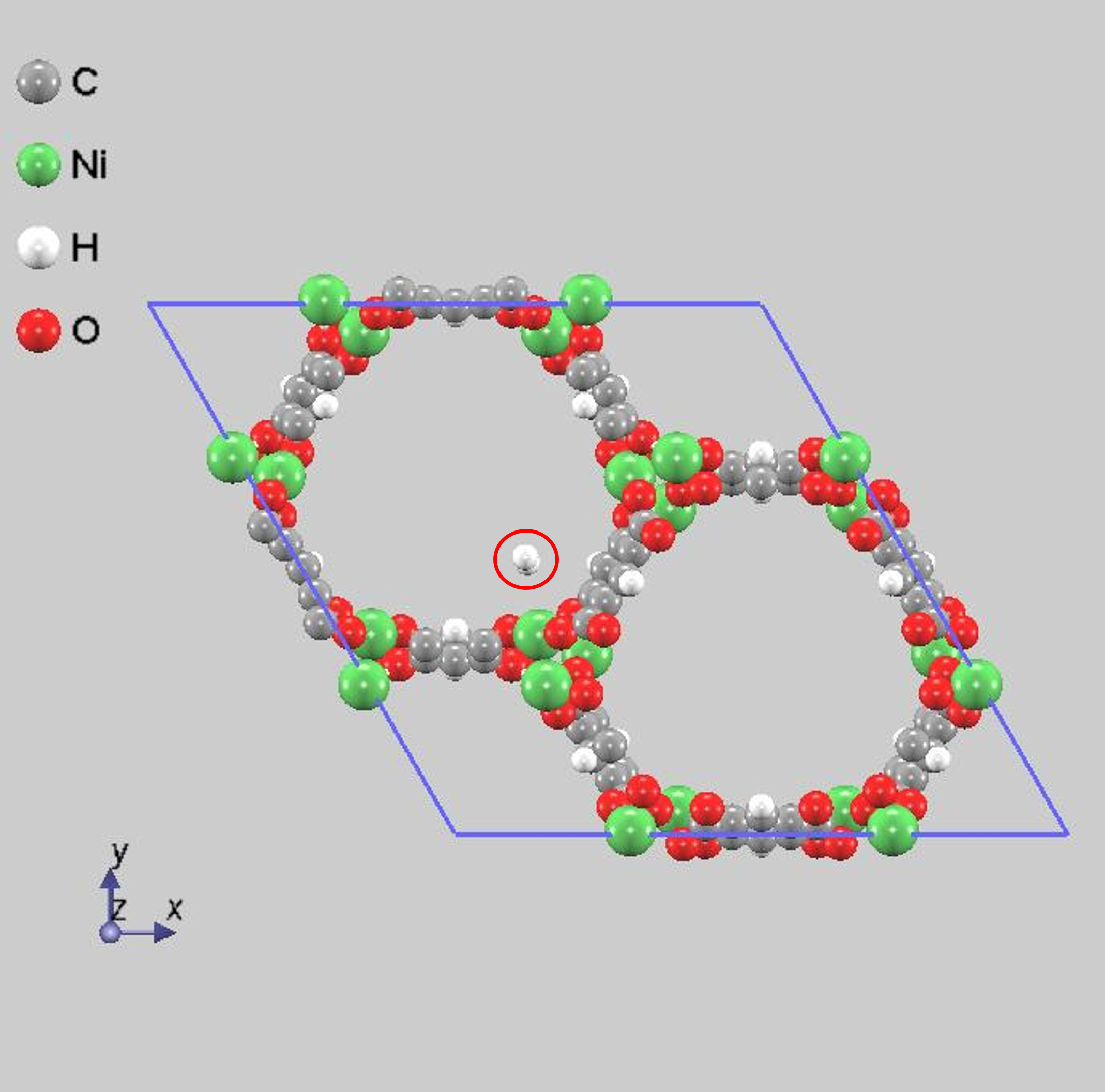
図2.1個の水素分子が吸着している場合の安定構造
表2.吸蔵された水素分子の特徴的な原子間距離
| H1-H2 [nm] | 0.076 |
|---|---|
| H1-Ni [nm] | 0.311 |
| H2-Ni [nm] | 0.335 |
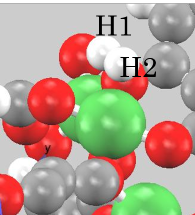
別の角度から見た水素吸着の拡大図
次に、すべてのオープンメタルサイトに水素分子が吸着している場合の構造最適化を行いました。計算セル内にオープンメタルサイトは18個の水素分子が吸蔵されていることになります。図3に結果を示します。吸蔵されている水素分子の水素原子間距離は、0.076 [nm]となっており、1個の水素分子が吸蔵されている場合と同様に、分子状で吸蔵されていることを意味します。水素原子とNi原子間の距離は、0.257~0.312 [nm]の間にあり、1分子吸着よりも近い距離に吸蔵されていました。これは、金属-水素の相互作用による効果と考えるよりも、フレーム内に多くの水素分子が存在することによる水素分子間の相互作用 (斥力) による影響であると考えられます。
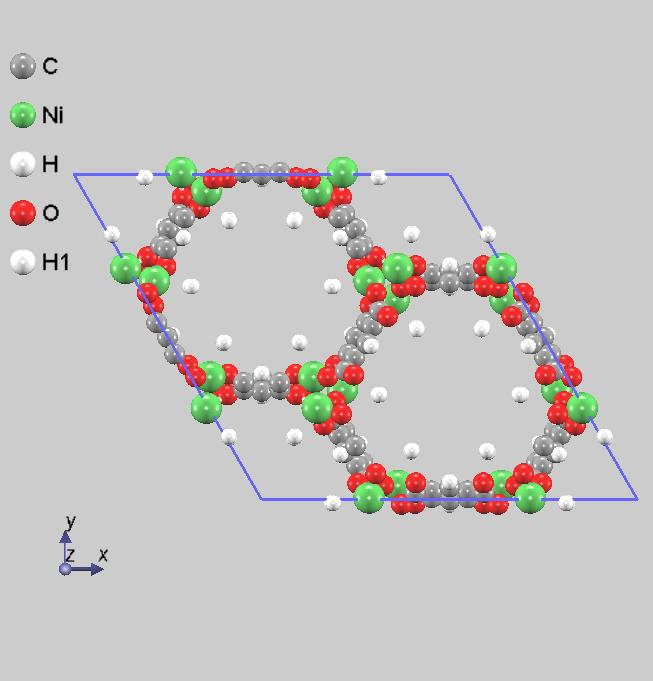
図3.すべてのオープンメタルサイトに水素分子が吸着している場合の安定構造
水素分子を吸蔵したNi2(m-dobdc)の電子状態や吸蔵エネルギーの解析#
水素分子とNi2(m-dobdc)の相互作用の様子を解析するために、吸蔵エネルギーと電子状態を解析しました。吸蔵エネルギー の定義は、
とします。ここで、 は水素を吸蔵しているMOFの全エネルギー、 は孤立水素分子の全エネルギー、 はMOFの全エネルギーです。また、 はMOFに吸蔵されている水素分子の計算セル当たりの数です。この定義から、 の値が負になれば、安定に吸蔵されることを示します。表3に結果を示します。先行研究 [1] では、-12.93 [kJ/mol]と報告されており、すべてのオープンメタルサイトに水素分子が吸着している場合には、先行研究に近い値が得られています。
表3.Ni2(m-dobdc)への水素分子吸着の吸蔵エネルギー
| \(E_{\text{occ}}\) [kJ/mol] | |
|---|---|
| 1分子の吸蔵 | -38.16 |
| 18分子の吸蔵 | -11.53 |
次に、相互作用の様子を調べるために、次の式で定義する差電子密度分布を調べました。
ここで、は水素を吸蔵しているMOFの価電子密度、は水素分子の価電子密度、はMOFの価電子密度です。この定義により、正の値は吸着により電子が増加していることを、負の値は吸着により電子が減少していることを示します。図4に1個のオープンメタルサイトに水素分子が吸着している場合を示します。
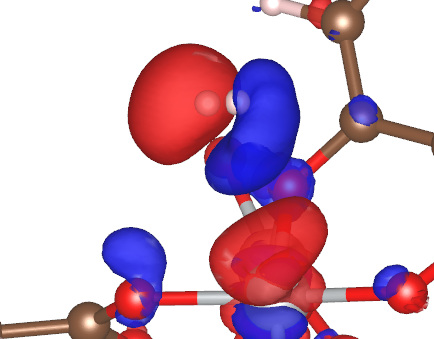
図4.1個のオープンメタルサイトに水素分子が吸着している場合の差電子密度分布の等密度面 (1.0×10-4 electrons/Bohr3)。赤が増加している領域、青が減少している領域。
図4は吸着により水素分子と金属原子の間に電子のやり取りがあるように見えます。しかし、この等密度面が1.0×10-4 electrons/Bohr3としているので、わずかな量の変化しかなく、化学的な結合はないと考えられます。また、この結果からわかることは、吸着により水素分子の一方の水素原子は電子が減少し、もう一方の水素原子は電子が増加するというようなヘテロな相互作用をしているということです。球近似で原子当たりの電子量を見積もると、Ni原子に近い水素(表2のH1)はわずかに電子数が増加し、遠い水素(表2のH2)はほとんど変化していないという結果になりました。この結果は、文献[1]のFigure 8の結果と一致していると考えられます。
また、吸蔵エネルギーに対して、van der Waals補正がどの程度影響しているかを検証するために、1個の水素分子が吸蔵されている場合について、DFT-D3の補正を考慮しないシミュレーションを実行しました。このときの吸蔵エネルギーは、-17.22 [kJ/mol]となりました。van der Waalsの相互作用を補正することにより、約21 [kJ/mol]の吸蔵エネルギーの増加がありました。この結果から、MOFへ水素分子が分子状態のまま吸蔵されるシミュレーションを行うには、van der Waals相互作用を考慮することが重要であることがわかりました。
まとめ#
第一原理計算ソフトウェアAdvance/PHASEを用いて、MOF(金属有機構造体)の一種であるNi2(m-dobdc)への水素分子の吸蔵シミュレーションを行いました。その結果、水素は原子に解離せず分子の状態で安定して吸蔵され、その吸蔵エネルギー(-11.53 kJ/mol)は先行研究の値とよく一致し、本計算手法の妥当性が確認されました。さらに電子状態の解析から、この吸蔵は強い化学結合ではなく、主に弱いファンデルワールス力に支配されていることが判明しました。このことから、MOFへの分子吸蔵シミュレーションにおいてはファンデルワールス力の補正が不可欠であり、本解析で検証された手法は他のMOF材料の評価にも応用可能であると結論付けられます。
本解析の詳細や、研究への適用可能性に関するご相談はこちら
お問い合わせ参考文献#
- M. T. Kapelewski et al., J. Am. Chem. Soc. 136 (2014) 12119.
- S. Grimme, J. Antony, S. Ehrlich, H. Krieg, J. Chem. Phys. 132 (2010) 154104.
関連ページ#
- 第一原理計算ソフトウェア Advance/PHASE
- 解析分野:ナノ・バイオ
- 産業分野:材料・化学