超分子電荷移動錯体C60(ferrocene)2の第一原理シミュレーション:構造・電子状態解析#

電子受容体(アクセプター)であるフラーレン(C60)と、電子供与体(ドナー)であるフェロセンは、共結晶化することでC60(ferrocene)2という超分子錯体を形成します。 この規則的なネットワーク構造の安定化には、両分子間に生じる電荷移動相互作用が深く寄与していると考えられています。本解析では、第一原理計算ソフトウェアAdvance/PHASEを用いて、C60(ferrocene)2の結晶構造最適化および電子状態解析を実施しました。DFTシミュレーションにより、X線回折(XRD)の実験パターンを精度よく再現でき、ドナー・アクセプター間におけるフロンティア軌道の分布、およびそれらの空間的重なり(軌道混成)が可視化できました。
Keywords: 第一原理計算, DFTシミュレーション、超分子錯体, 電荷移動錯体, フラーレン, フェロセン, 構造最適化, X線回折(XRD), HOMO, LUMO, 部分電荷密度
計算モデルと計算条件#
初期構造には、文献 [1] に報告された低温構造のX線結晶構造解析データ(CIF)を使用しました。この結晶構造は三斜晶系(空間群 P-1)であり、単位胞内に1分子のC60と2分子のフェロセンを含みます。1つのC60分子の周囲にフェロセンがインターカレートするような配置をとっており、フェロセン分子のシクロペンタジエニル環がC60の五員環面と平行になるようにパッキングされています(図1)。
第一原理計算において、分子間におけるファンデルワールス力(分散力)を適切に評価するため、交換相関汎関数にはGGA (PBE) に加えて分散力補正(DFT-D3)を導入し、原子位置とセルサイズの大域的な構造最適化を行いました。k点サンプリングには3x3x3 k点メッシュを使用しました。
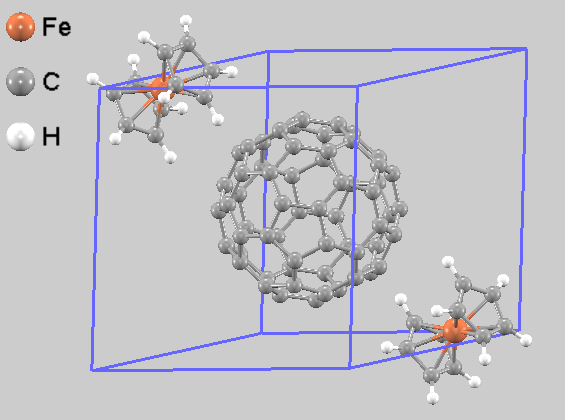
図1. 最適化されたC60(ferrocene)2の計算モデル(単位胞)。
計算結果と考察#
格子定数と構造最適化#
X線回折(XRD)実験では、散乱体である水素原子の電子雲が炭素側に強く偏るため、C-H結合長が実態の原子間距離より短く観測されるという原理的課題があります。本計算において第一原理に基づく構造緩和を行った結果、C-H結合長は物理的に妥当な値(1.082~1.083 Å)になることが確認されました。得られた最適化後の格子定数は表1の通りであり、文献で報告されている実験値(143 K)[1] と良好な一致を示しました。これは、分子性結晶において分散力補正を含めた本計算手法の妥当性を裏付けています。
表1. C60(ferrocene)2の格子定数の比較
| a (Å) | b (Å) | c (Å) | α (°) | β (°) | γ (°) | |
|---|---|---|---|---|---|---|
| 計算値 | 10.018 | 10.441 | 11.419 | 95.31 | 90.89 | 118.05 |
| 実験値 (143 K) | 9.899 | 10.366 | 11.342 | 95.55 | 90.96 | 118.33 |
X線回折(XRD)パターンの検証#
最適化された結晶構造からXRDパターン(CuKα線)をシミュレーションし、文献 [2] に報告されている実験データとのクロスチェックを行いました。 その結果、実験データにおいて最大強度を示す 2θ = 19°〜19.5° 付近の強力な回折ピークの位置を正確に捉えているだけでなく、低角側(11°〜13.5°)に見られる複数の特徴的なピーク群や、17°〜21° にかけて現れるピークの分裂および相対強度バランスについても、実験結果の傾向を非常によく再現しました。この結果は、シミュレーションで得られた大域的安定構造が、実際の結晶構造を高精度に再現できていることを証明しています。
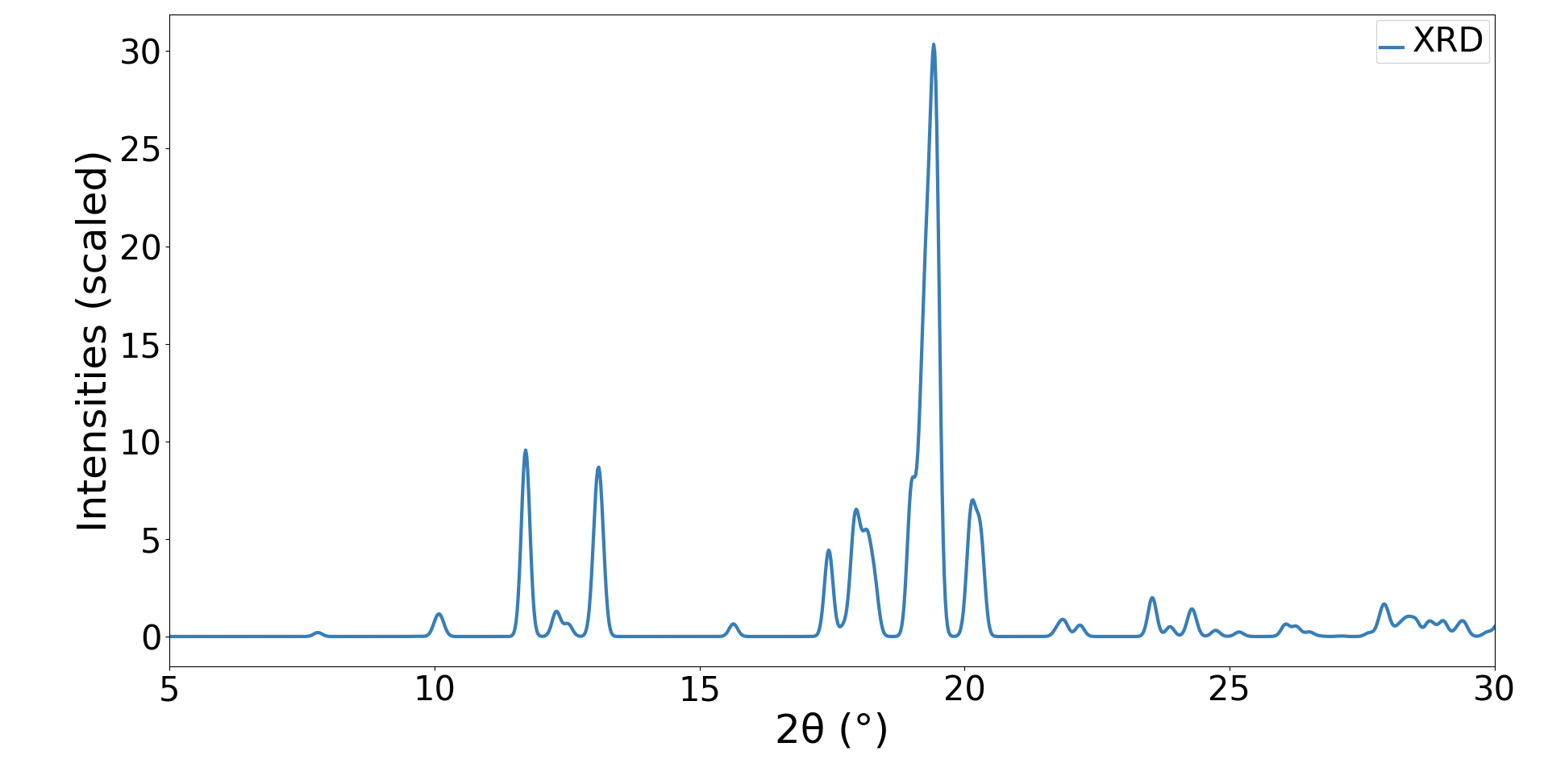
図2. C60(ferrocene)2のXRDシミュレーション結果。
電子状態解析:価電子帯上端(VBM)近傍#
ドナー側の電子状態を評価するため、VBM近傍(-0.5 eV 〜 0 eV)の部分電荷密度を計算しました(図3左)。図中で黄色く示された等値面は、フェロセンの中心金属であるFe原子の周囲でシクロペンタジエニル環と平行に広がる分布と、上下の環の方向へ伸びた2つのローブを持つ形状を示しました。また、Γ点で最高被占有軌道(HOMO)の波動関数を可視化すると、フェロセンのFe原子上に「四つの葉」の形をした明確なd軌道由来の分布が現れます(図3右)。これらの結果は、VBM(HOMO)が主にフェロセンのFe 3d軌道から構成されていることを明確に示しています。
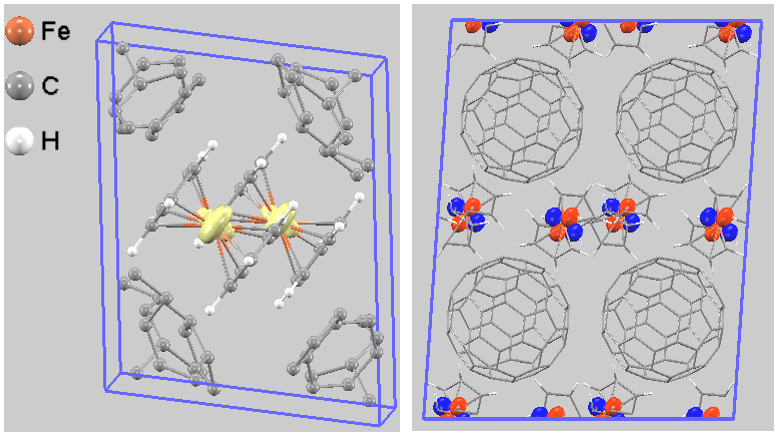
図3. (左)VBM近傍(-0.5 eV 〜 0 eV)の部分電荷密度。(右)Γ点でのHOMOの波動関数の分布(視認性のため2x2x2スーパーセル使用)。
電子状態解析:伝導帯下端(CBM)近傍#
一方、アクセプター側で電子の受け皿となるCBM近傍(0 eV 〜 0.5 eV)の部分電荷密度を計算すると、その分布の大部分はC60分子全体を包み込むように存在しています(図4左)。 また、Γ点での最低空軌道(LUMO)の波動関数を可視化すると、主にC60のt1u軌道からなることを示しています(図4右)。
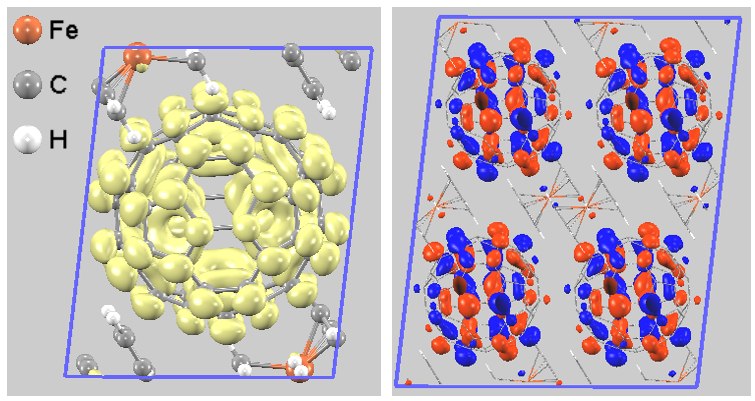
図4. (左)CBM近傍(0 eV 〜 0.5 eV)の部分電荷密度。(右)Γ点でのLUMOの波動関数の分布(視認性のため2x2x2スーパーセル使用)。
ドナー・アクセプター間の軌道混成の可視化(低スケール表示)#
フラーレンとフェロセン間の電子的相互作用(電荷移動)の本質に迫るため、部分電荷密度の等値面レベル(表示スケール)をさらに下げて電子分布を詳細に解析しました。
VBM近傍(HOMO帯)の分布において、フェロセン側だけではなく、C60側にも明確な分布が観察されます(図5左)。同時に、CBM近傍(LUMO帯)の電荷密度においても表示の閾値を下げると、アクセプターであるC60分子上の分布に加えて、ドナーであるフェロセンのFe原子上に明確なd軌道由来の分布が現れます(図5右)。
このように、LUMO帯域にフェロセンのd軌道成分が、HOMO帯域にC60の成分が互いにわずかに混じり合う(ブレンドされる)現象は「軌道混成(Orbital Hybridization)」と呼ばれます。 これは、本系が単なる分子の物理的な寄せ集めではなく、ドナーのd軌道とアクセプターのπ*軌道間に電子が移動するための「架け橋(軌道の重なり)」が形成されていることを意味します。この軌道混成の可視化は、C60(ferrocene)2が真の電荷移動超分子錯体であることを鮮明に示しています [2]。
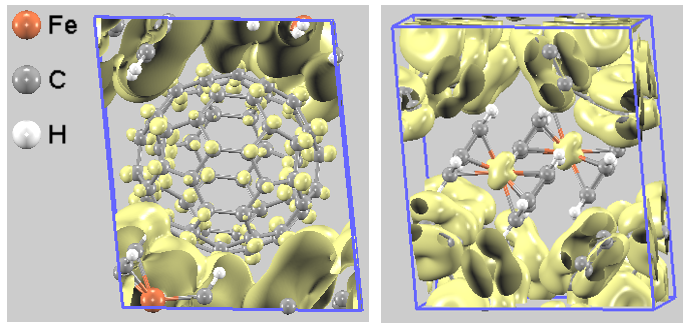
図5. より低いスケール(閾値)で表示した部分電荷密度。左:VBM近傍、右:CBM近傍。
【補足】 電荷移動励起エネルギーの定量評価について#
本事例ではGGA(PBE汎関数)を用いて構造最適化や電子状態の解析を行いましたが、基底状態におけるHOMO-LUMOのエネルギー差から、電荷移動(CT)励起エネルギーを直接見積もるような定量評価については留意が必要です。空間的に離れたドナー・アクセプター間で生じるCT励起エネルギーは、標準的なLDAやGGA汎関数を用いると、自己相互作用誤差等の原理的な問題により著しく過小評価されることが広く知られています。
実際に文献 [2] の研究では、このCT励起エネルギーをより正確に見積もるため、Slater遷移(ST)法や時間依存密度汎関数法(TD-DFT)に基づく修正線形応答(MLR)スキーム [3] といったアプローチが採用されています。実験的に観測されたCT吸収帯(782 nm付近、約1.59 eV)に対し、MLR法による計算値(1.016 eV)は吸収端(約1000 nm、1.24 eV)に対応するものとして合理的に解釈されています。なお現在、本系のような結晶系(周期境界条件)でCT励起エネルギーのより定量的な比較・予測を行う際には、長距離補正汎関数(LC-ωPBE [4] など)が有力なアプローチとなります。
まとめ#
第一原理計算ソフトウェアAdvance/PHASEを用いたC60(ferrocene)2のDFTシミュレーションにより、XRDの実験プロファイルを高精度に再現する最適化構造が得られました。また、部分電荷密度およびフロンティア軌道の可視化を通じて、HOMOがフェロセンのFe 3d軌道に、LUMOが主にC60のt1u軌道に局在していることを確認しました。さらに、低スケールでの部分電荷密度解析により、両者の間に軌道混成(電荷移動相互作用)が生じている様子を視覚的かつ物理的に捉えることができました。これらの成果は、超分子錯体における電子物性やドナー・アクセプター相互作用の解明において、第一原理計算が強力なアプローチであることを示しています。
本解析の詳細や、研究への適用可能性に関するご相談はこちら
お問い合わせ参考文献#
- J. D. Crane, P. B. Hitchcock, H. W. Kroto, R. Taylor, and D. R. M. Walton, "Preparation and Characterisation of C60(ferrocene)2", J. Chem. Soc., Chem. Commun. 1992, 1764 (1992).
- T. Wakahara, M. Sathish, K. Miyazawa, C. Hu, Y. Tateyama, Y. Nemoto, T. Sasaki, and O. Ito, "Preparation and Optical Properties of Fullerene/Ferrocene Hybrid Hexagonal Nanosheets and Large-Scale Production of Fullerene Hexagonal Nanosheets", J. Am. Chem. Soc. 131, 9940 (2009).
- C. Hu, O. Sugino, and Y. Miyamoto, "Modified linear response for time-dependent density-functional theory: Application to Rydberg and charge-transfer excitations", Phys. Rev. A 74, 032508 (2006).
- O. A. Vydrov and G. E. Scuseria, "Assessment of a long-range corrected hybrid functional", J. Chem. Phys. 125, 234109 (2006).
関連ページ#
- 第一原理計算ソフトウェア Advance/PHASE
- 解析分野:ナノ・バイオ
- 産業分野:材料・化学