擬ポテンシャル法によるEELS/XAFSの絶対エネルギー位置とスペクトル形状の算出#

電子エネルギー損失分光法(EELS/ELNES)やX線吸収分光法(XAFS/XANES)は、物質の局所的な電子状態や原子構造を解析するための強力な手法です。 第一原理計算はこれらのスペクトル解釈に不可欠ですが、従来の平面波基底を用いた擬ポテンシャル法では、内殻電子を陽に扱わないため、スペクトルの「絶対エネルギー位置(閾値)」を直接算出することが困難でした。そのため、実験スペクトルに合わせて計算結果を手動でシフトさせる手法が一般的でした。本事例では、第一原理計算ソフトウェアAdvance/PHASEを用い、Core-hole(内殻空孔)を導入した励起擬ポテンシャルと SCF法を組み合わせることで、MgOおよび3C-SiCのK吸収端スペクトルにおける絶対エネルギー位置と形状を高精度に算出した結果を紹介します。
Keywords: 第一原理計算, DFTシミュレーション, EELS/XAFS, ELNES/XANES, K吸収端(K-edge), Core-hole擬ポテンシャル, SCF法, 化学シフト
理論背景:絶対エネルギー位置の算出手法#
1. SCF法による遷移エネルギーの定義#
全電子法とは異なり、平面波擬ポテンシャル法では価電子のみを計算対象とするため、遷移エネルギー()を単純な全エネルギー差から求めることができません。本解析では、Projector Augmented Wave (PAW)法をベースに、溝口ら [1] によって提案された手法に基づき、結晶系でのエネルギー差に孤立原子計算による補正項を加えることで、絶対値としての遷移エネルギー を算出しました。
2. 各エネルギー項の定義と交換相関エネルギーの取り扱い#
Advance/PHASEでは、以下のスキームを用いて各項を計算します。
-
(結晶系のエネルギー差): ここで、 はCore-hole擬ポテンシャルを用いた遷移状態の全エネルギー、 は通常の擬ポテンシャルを用いた基底状態の全エネルギーです。
-
(内殻補正項): 孤立原子における全電子計算(All-electron)から得られる、励起状態と基底状態の全エネルギー差から、価電子の寄与を差し引いたもの。
厳密には、交換相関エネルギー(\(E_\text{xc}\))は非線形な性質を持つため、内殻成分と価電子成分の単純な和として表すことができません(\(E_\text{xc}(\text{all}) \neq E_\text{xc}(\text{core}) + E_\text{xc}(\text{val})\))。
そのため、NLCC(非線形内殻補正)を用いた擬ポテンシャル計算では、結晶系の全エネルギー(\(E_\text{ground}^\text{solid}\) および \(E_\text{excited}^\text{solid}\))に、内殻と価電子の相互作用を含んだ交換相関エネルギーが既に正しく含まれています。したがって、エネルギーの二重カウントを防ぐため、単原子補正項(\(\Delta E_\text{core(atom)}\))の算出時には、交換相関エネルギー項を除外する処理を行っています。これにより、擬ポテンシャル法においても矛盾なく正しい遷移エネルギーを定義しています。
Pythonスクリプトによる解析ワークフローの効率化#
第一原理計算の結果から最終的なスペクトル図を得るまでのプロセスは、複数の出力ファイルからのデータ抽出と複雑な演算を伴います。本事例では、Pythonスクリプトを活用することで、以下の処理を自動化・効率化し、スムーズな解析を実現しました。
1. データの自動抽出と演算#
Advance/PHASEの出力ファイルから、基底状態・励起状態それぞれの全エネルギー、および単原子計算の結果を自動的に抽出し、上述のSCF理論式に基づいて絶対エネルギーシフト量(閾値)を算出します。表1には、MgO (O K-edge) および3C-SiC (C K-edge) の絶対エネルギーシフト量を示しています。
表1. Python処理により算出されたシフト量
| 物質 / 吸収端 | 算出されたシフト量 (eV) |
|---|---|
| MgO (O K-edge) | 540.6 |
| 3C-SiC (C K-edge) | 289.0 |
2. スペクトル処理と可視化#
EELS/XAFS計算で得られた相対エネルギースペクトル(onset=0 eV)に対し、算出された絶対エネルギーシフト量を適用し、実験データと直接比較可能な形式でプロットを作成します。
解析結果と考察#
1. MgO O K-edgeの評価#
図1に、SCF法によるエネルギー補正を適用したMgOのEELS/XAFSスペクトルを示します。
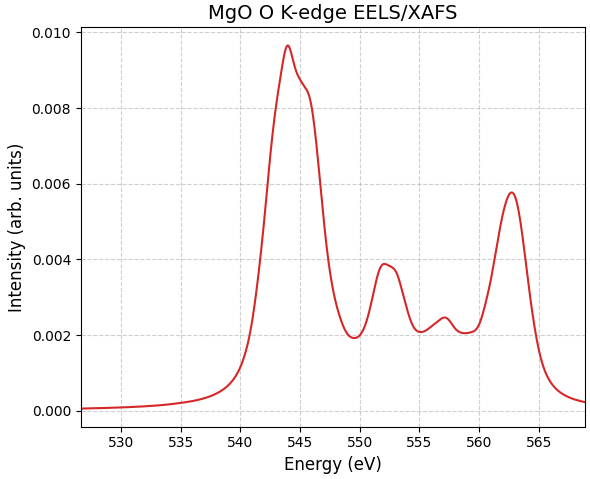
図1. Advance/PHASEを用いて計算されたMgOのO K吸収端スペクトル。横軸は算出された絶対エネルギー値で表示されています。
MgOのスペクトルでは、計算による吸収端の立ち上がりが 540.6 eV、第1ピーク位置は約544 eV と算出されました。Lindnerら [2] による実験結果(立ち上がり532 eV付近、第1ピーク位置は535 eV付近)と比較すると、絶対エネルギー位置は約9 eV程度高く見積もられています。この傾向は先行研究とも一致しており、例えば溝口ら [1] は TiO2 などの酸化物において、本手法を用いても数eV程度の誤差(過大評価)が生じることを報告しています 。 したがって、この系統的なエネルギーシフトは計算手法に内在する特性であると考えられ、相対的なスペクトル形状の再現性には影響しません。
しかし、立ち上がり位置(Onset)を基準としてスペクトル形状を比較すると、ピークの分裂やそれぞれの特徴的な構造が非常に良く再現されていることがわかります。これは、本手法が内殻励起に伴う電子緩和効果や、O原子周辺の局所構造・結合状態を適切に記述できていることを示しています。
2. 3C-SiC C K-edgeの評価#
図2に、SCF法によるエネルギー補正を適用した3C-SiCのEELS/XAFSスペクトルを示します。
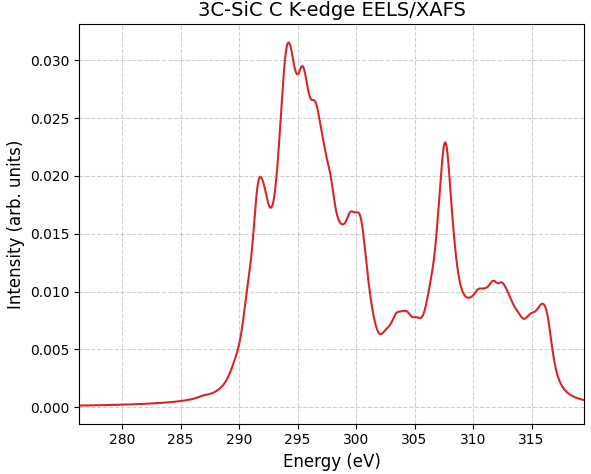
図2. Advance/PHASEを用いて計算された3C-SiCのC K吸収端スペクトル。横軸は算出された絶対エネルギー値で表示されています。
3C-SiCのスペクトルにおいては、吸収端が 289.0 eV、第1ピーク位置は292 eV と算出されました。Pedioら [3] の実験スペクトル(NEXAFS)では、立ち上がりが約283 eV、それに続く吸収端近傍の構造(ショルダー)が286 eV付近に観測されています。MgOと同様に、計算値は実験値に対して約6 eV程度の正のシフト(過大評価)が見られます。
絶対値のずれはあるものの、スペクトル全体の概形は実験結果とよく一致しています。特に、ピーク構造 の相対的なエネルギー間隔や強度比は、実験の特徴を正確に捉えています。Pedioらはこれらの特徴がC 2pおよびC 2sの空状態密度を反映していると報告しており、本計算はその電子状態の特徴を正しく再現できています。
3. 本手法の有用性と実際の材料解析への応用事例#
従来、擬ポテンシャル法を用いた計算では、スペクトルの横軸は相対値でしか議論できず、実験値との比較には「ピーク位置合わせ」という手動的な操作が必要でした。本解析で示したように、SCF法を用いることで、実験値に頼らずに絶対エネルギー位置を算出可能です。計算値には数eV~10eV程度の過大評価が含まれる傾向がありますが、このシフト量は系によらず比較的安定的です。
この「安定的なシフト量」と「高いスペクトル形状再現性」を組み合わせることで、未知の材料や複雑な界面構造における化学シフトの同定や、元素の配位環境(化学結合状態)の特定がより確実に行えるようになります。
実際に、Advance/PHASEを用いた本手法は、複雑な未知材料の同定において威力を発揮しています。例えば、文献 [4] では、リチウムイオン電池のSi負極上に形成されるSEI(固体電解質界面)被膜の解析において本手法を適用しました。この論文では、C K吸収端の計算スペクトルと実験スペクトルを比較することで、SEI中の高分子成分が「架橋型Poly(FEC)」であることを突き止めました。この際、C-F結合や架橋構造に由来する微細なピーク分裂( feature)の形状や相対強度が、計算によって正確に再現されたことが構造決定の決め手となりました [4]。
よって、絶対エネルギー位置に系統的なシフトが含まれる場合でも、それを補正してスペクトル形状に着目することで、実験だけでは困難な化学構造の特定が可能であることを実証しています。
まとめ#
本事例では、第一原理計算ソフトウェア Advance/PHASE を用いて、MgOおよび3C-SiCの内殻励起スペクトル(EELS/XAFS)を計算しました。Core-holeを考慮した励起擬ポテンシャルとSCF法に基づくエネルギー解析を連携させることで、スペクトル形状だけでなく、絶対エネルギー位置(閾値)を高精度に算出することが可能になっています。MgOのO K-edgeは約540.6 eV、3C-SiCのC K-edgeは約289.0 eVという、系統的なシフトを考慮すれば実験事実とよく整合する結果が得られました。本手法は、実験スペクトルの帰属や、微量添加元素・格子欠陥周辺の電子状態解析において有効なツールとなります。
本解析の詳細や、研究への適用可能性に関するご相談はこちら
お問い合わせ参考文献#
- T. Mizoguchi, I. Tanaka, S.-P. Gao, and C. J. Pickard, "First-principles calculation of spectral features, chemical shift and absolute threshold of ELNES and XANES using a plane wave pseudopotential method", J. Phys.: Condens. Matter 21, 104204 (2009).
- Th. Lindner, H. Sauer, W. Engel, and K. Kambe, "Near-edge structure in electron-energy-loss spectra of MgO", Phys. Rev. B 33, 22 (1986).
- M. Pedio, A. Giglia, N. Mahne, S. Nannarone, S. Giovannini, C. Cepek, F. Boscherini, R. Carboni, M. Benfatto, and S. Della Longa, "C K-Edge NEXAFS of 6H-SiC and 3C-SiC Systems", Physica Scripta T115, 308 (2005).
- Y. Kamikawa, K. Amezawa, and K. Terada, "Energy-Loss Near-Edge Structures and Low-Loss Structures of Polymers in a Solid Electrolyte Interface Formed from Fluoroethylene Carbonate on a Si Anode Clarified by DFT Calculations", J. Phys. Chem. C 125, 3890 (2021).
関連ページ#
- 第一原理計算ソフトウェア Advance/PHASE
- 解析分野:ナノ・バイオ
- 産業分野:材料・化学