多孔性金属錯体DMOF-1の第一原理シミュレーション:構造物性と電子状態解析#

金属イオンと有機物(リンカー)から構成される金属有機構造体(Metal Organic Framework, MOF)は、その規則正しいナノ多孔性を活かし、ガスの吸着・貯蔵・分離材料として注目を集めています。特にDMOF-1は、ゲスト分子に応じて構造が柔軟に変化するユニークな特性で知られています。本解析では、第一原理計算ソフトウェアAdvance/PHASEを用いて、多孔性金属錯体DMOF-1の結晶構造と電子状態を解析し、その物性が発現するメカニズムの解明を目指します。
Keywords: 第一原理計算 (DFT), 金属有機構造体(MOF), DMOF-1, 構造最適化, X線回折(XRD), 電荷密度, HOMO, 構造柔軟性
計算モデルと計算条件#
本解析で用いた計算モデルは、pcuというトポロジーを持つDMOF-1(化学式: [Zn2(terephthalate)2(DABCO)])です。この物質は、以下の3つのビルディングブロックが自己集合して結晶構造を形成します。
- 金属イオン: 亜鉛 (Zn2+)
- リンカー: テレフタル酸 (BDC)
- ピラー(柱)リンカー: 1,4-ジアザビシクロ[2.2.2]オクタン (DABCO)
構造的には、亜鉛イオンとテレフタル酸が2次元のシートを形成し、そのシート間をDABCO分子が柱(ピラー)のように連結することで、3次元的なジャングルジム様の骨格とナノサイズの細孔(チャンネル)が形成されます(図1)。
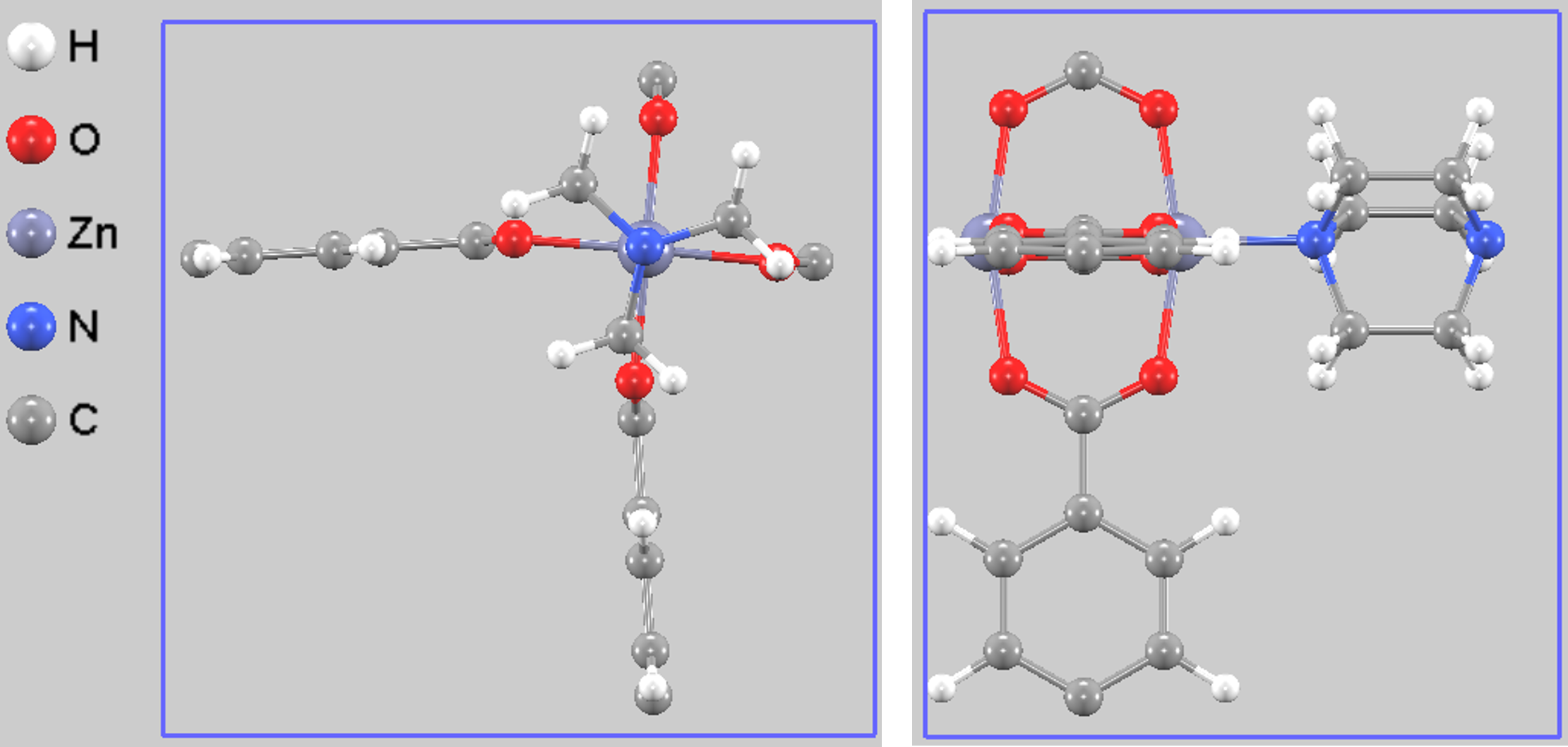
図1. 最適化されたDMOF-1の計算セル(単位胞)。左:上面図、右:側面図。
本解析で用いた主な計算条件は表1に示されています。交換相関汎関数にはGGA (PBE) を採用し、MOF特有の多孔質構造や分子間の相互作用を精密に評価するため、ファンデルワールス力を考慮したDFT-D3による分散力補正を併用しました。なお、ストレステンソルを用いた計算では、カットオフエネルギーをさらに2倍に設定しました。
表1. 計算条件の概要
| 項目 | 設定 |
|---|---|
| 擬ポテンシャル | ウルトラソフト擬ポテンシャル |
| 交換相関汎関数 | GGA (PBE) + DFT-D3 |
| 波動関数のカットオフエネルギー | 30 Rydberg(エネルギー計算) |
| k点サンプリング | 1x1x2 |
計算結果と考察#
構造最適化#
第一原理計算に基づきDMOF-1の結晶構造を最適化した結果、格子定数は a=b=10.929 Å, c=9.570 Å となりました。この値は、文献 [1] で報告されている実験値 (a=b=10.929 Å, c=9.608 Å) と非常によく一致しており、本計算手法の妥当性が確認されました。
X線回折(XRD)パターンの比較#
計算で得られた最適化構造からシミュレーションしたXRDパターン (CuKa, λ = 1.54184 Å, smearing: 0.05 eV)と、実験 [2] で測定されたXRDパターンを比較しました。図2に示すように、回折ピークが現れる角度(2θ)と相対的な強度の両方において、計算結果と実験結果は非常によい一致を示しています。特に、約8.1°に現れる最も強度の高いピークをはじめ、他の主要なピークも正確に再現されており、計算によって得られた原子配置の妥当性を裏付けています。
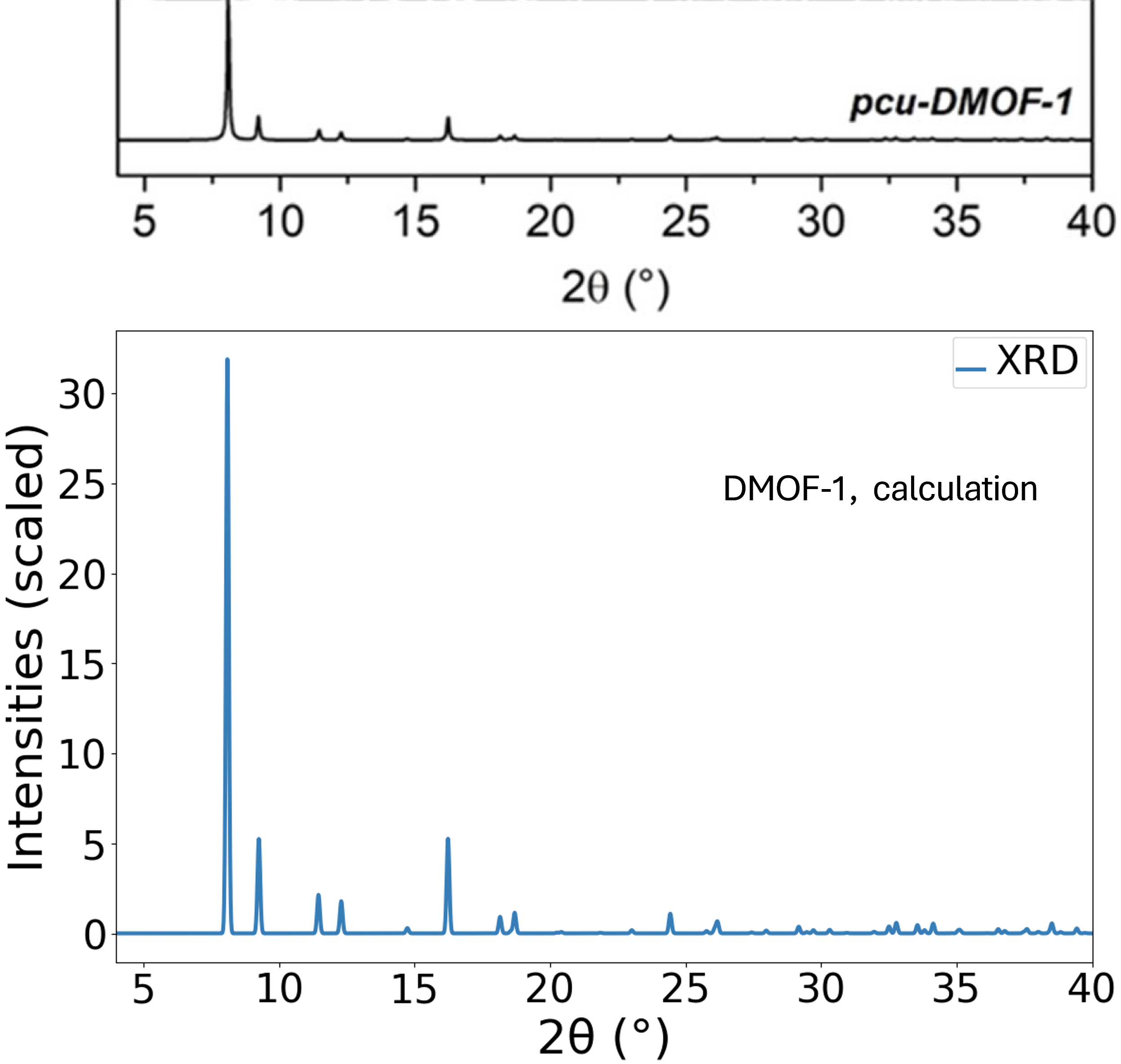
図2. DMOF-1のXRDパターンの比較。(上) 実験測定結果、(下) 本計算によるシミュレーション結果。
電子状態の解析#
全電荷密度と構造の柔軟性#
図3は、DMOF-1結晶内の全電荷密度を可視化したものです。黄色で示された等値面は電子が密に存在する領域を示しており、原子間の化学結合の様子を明確に捉えることができます。等値面を表示する閾値を高く設定すると、図4に示すように、電荷密度が特に高い領域(オレンジ色)が亜鉛(Zn)原子と酸素(O)原子の結合部分に集中して現れます。
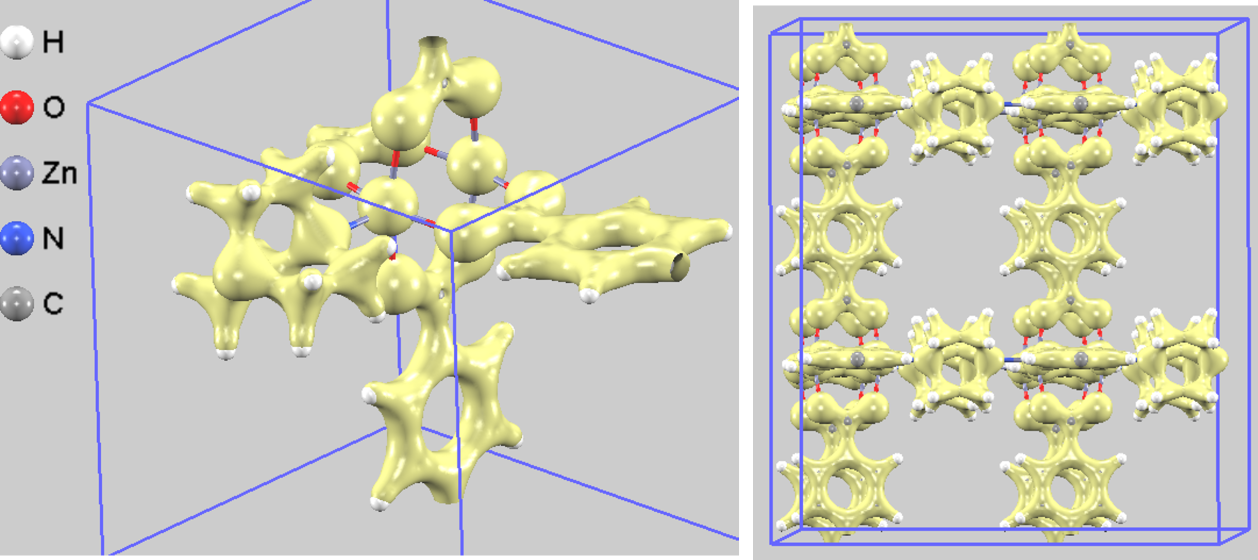
図3. DMOF-1の全電荷密度。左:単位胞、右:視認性を高めた2x2x2スーパーセル表示。
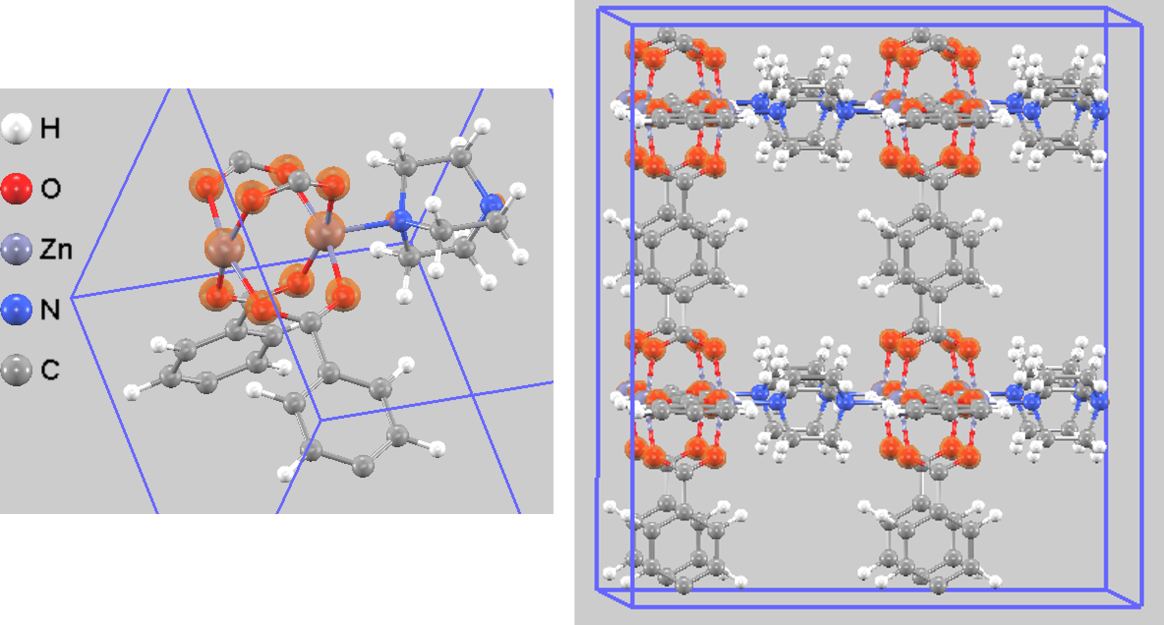
図4. DMOF-1の全電荷密度(高い閾値)。左:単位胞、右:視認性を高めた2x2x2スーパーセル表示。電荷密度がZn-O結合周りに局在しています。
この電子分布の不均一性は、DMOF-1が持つユニークな「柔軟性」の起源を理解する上で重要な手がかりとなります [3, 4]。電荷密度が高くイオン結合性の強いZn-Oクラスター部分は、物理的に変形しにくい「剛直な節(ノード)」として振る舞うと考えられます。一方で、電荷密度が相対的に低い有機リンカーの部分は、結合周りの回転などが比較的容易な「柔軟な梁(はり)」としての役割を担っていると解釈できます。
このように、性質の異なる「剛直な節」と「柔軟な梁」が三次元的に組み合わさった構造であることが、DMOF-1が結晶性を保ったまま外部刺激に応答して骨格全体を大きく変形させるという、特異な物性につながっていると示唆されます。
部分電荷密度(HOMO)と機能性#
物質の反応性を探る上で重要なフロンティア軌道である最高被占有軌道(HOMO)の電子分布を解析しました(図5)。その結果、HOMOに由来する電子密度は、フレームワーク内で最も反応性が高い(電子を供与しやすい)領域が、亜鉛イオン(Zn2+)とそれに配位する酸素原子(O)の周辺であることを示しています。
この電子の局在は、DMOF-1の機能性と密接に関連する可能性があります [3, 4]。電荷が分極したZn-Oサイトは、細孔表面においてゲスト分子と相互作用する活性点になると考えられます。特に、二酸化炭素(CO2)のように部分的に正の電荷を帯びた原子を持つ分子に対しては、有効な吸着サイトとして機能する可能性があり、これがDMOF-1のガス吸着特性の一因となっていると推測されます。このように、電子状態の解析は、MOFの機能発現メカニズムを解明するための重要な知見をもたらします。
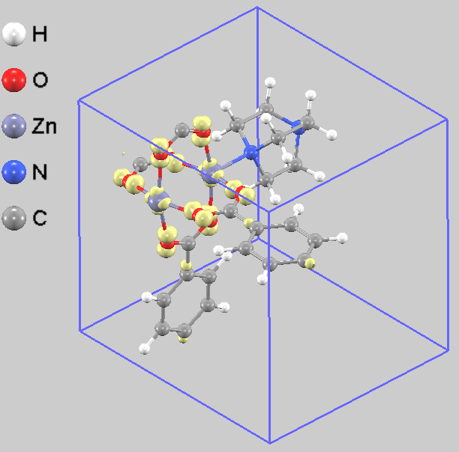
図5. DMOF-1のHOMOに由来する部分電荷密度。
まとめ#
本解析では、第一原理計算ソフトウェアAdvance/PHASEを用いて多孔性金属錯体DMOF-1の物性解析を行いました。計算による結晶構造とXRDパターンは実験結果と非常によく一致しました。電子状態解析からは、電荷密度がZn-Oの配位結合部分に集中することが明らかになり、これが構造の「剛直な節」として機能し、有機リンカーの「柔軟な梁」と組み合わさることで、DMOF-1特有の構造柔軟性が発現するメカニズムが示唆されました。また、HOMO電子も同領域に局在しており、これが選択的なガス吸着機能の起源であることを示唆しました。本事例は、第一原理計算がMOF材料の特性を深く理解し、新たな機能性材料を設計開発する上で強力なツールであることを示しています。
本解析の詳細や、研究への適用可能性に関するご相談はこちら
お問い合わせ参考文献#
- D. N. Dybtsev, H. Chun, and K. Kim, "Rigid and flexible: a highly porous metal–organic framework with unusual guest-dependent dynamic behavior", Angewandte Chemie International Edition 43, 5033 (2004).
- D. Sensharma and S. M. Cohen, "Ligand cross-links as a design element in oligo-and polyMOFs", Chemical Science 15, 20448 (2024).
- S. Kitagawa, R. Kitaura, and S. I. Noro, "Functional porous coordination polymers", Angewandte Chemie International Edition 43, 2334 (2004).
- 北川進、“多孔性金属錯体の合成と機能に関する研究”、Bulletin of Japan Society of Coordination Chemistry 51, 13 (2008).
関連ページ#
- 第一原理計算ソフトウェア Advance/PHASE
- 解析分野:ナノ・バイオ
- 産業分野:材料・化学