c-BN(001)表面における窒素ダイマー再構成の第一原理MDシミュレーション#

立方晶窒化ホウ素 (c-BN) は、ダイヤモンドに次ぐ高い硬度を持つ超硬材料であり、高温での安定性にも優れています。その特性から、焼入れ鋼などの難削材を加工するための切削工具材料としてすでに広く実用化されています。さらに、その大きなバンドギャップを活かした次世代のパワーデバイスや深紫外発光デバイスの材料としても活発な研究開発が進められています。これらの応用において、c-BNの表面構造や安定性は材料の性能を左右する重要な因子です。特にc-BN(001)表面は、終端原子の種類によって複雑な再構成構造をとることが知られています。本解析では、第一原理計算ソフトウェアAdvance/PHASE を用い、c-BN(001)表面のN終端構造が安定なN-Nダイマー(二量体)を形成する過程を、第一原理分子動力学(MD)シミュレーション によって再現し、その構造安定性を詳細に解析します。
Keywords: 第一原理計算 (DFT), 分子動力学 (MD), c-BN(001), 表面再構成, N-Nダイマー, スラブモデル
計算モデルと計算条件#
バルク格子定数の決定#
まず、表面モデルを構築するために、c-BNバルク結晶の格子定数を第一原理計算によって決定しました。図1に示すように、結晶の体積を様々に変化させて全エネルギーを計算し、エネルギー-体積(E-V)曲線を求めました。この曲線をフィッティングすることで、c-BNの平衡格子定数 a = 3.630 Å、体積弾性率 B = 364.5 GPaという値が得られました。これらの計算値は、実験値 [1](a = 3.615 Å, B = 369 GPa) と非常に良好な一致を示しており、本計算で用いる擬ポテンシャルや計算条件の妥当性が確認されました。
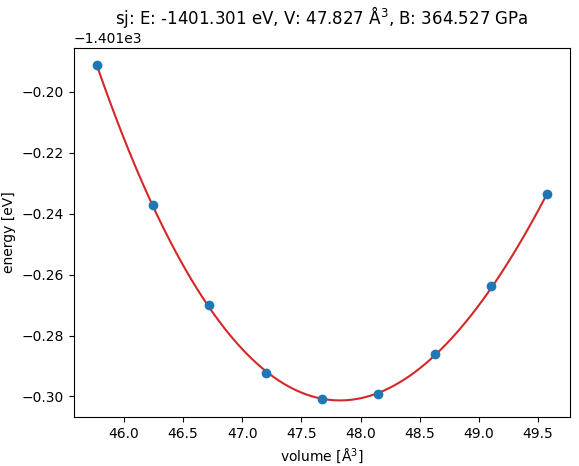
図1. c-BNバルク結晶のエネルギー-体積曲線。計算から得られた平衡格子定数と体積弾性率は実験値とよく一致しています。
スラブモデルの構築と計算条件#
MDシミュレーションの初期構造として、上記で決定した格子定数を用い、バルク結晶から切り出したc-BN(001) 2x1表面のスラブモデルを構築しました。このモデルはN終端表面を想定しています。スラブモデルの最下層のB原子は、バルク内部の結合を模擬するため、電子数を1.25個に設定した擬水素(H)原子で終端しました。これは、c-BN表面の計算で実績のある手法です [2]。MDシミュレーション中、この最下層のB原子と終端の擬水素原子の座標は固定し(図2で正常色)、それより上層の原子(図2で濃い色)は自由に動けるように設定しました。
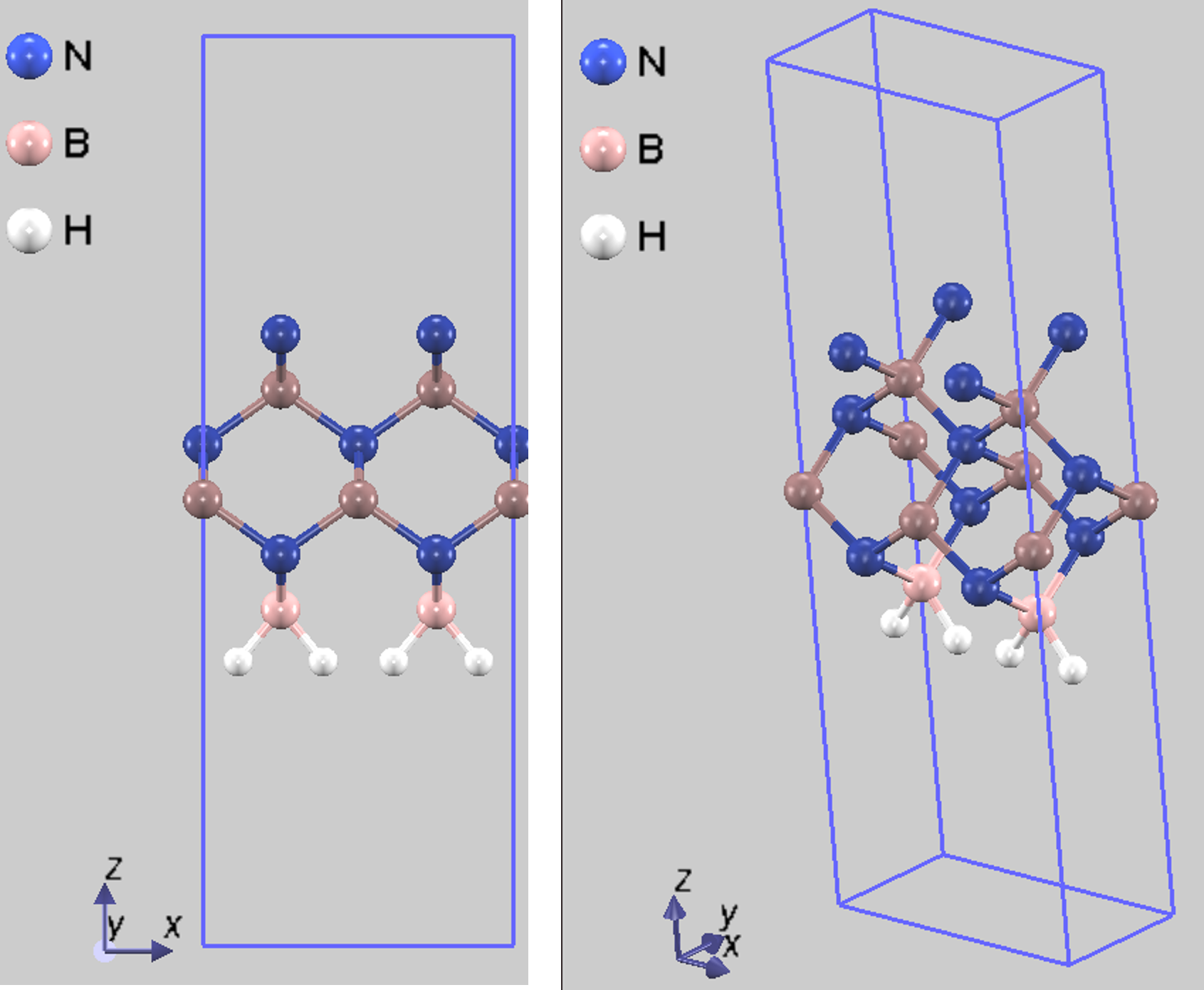
図2. MDシミュレーションに用いたc-BN(001) 2x1表面スラブモデルの初期構造(側面図および斜視図)。Hは擬水素原子です。濃色で示す原子が可動層、淡色で示す原子が固定層です。
本解析で用いた主な計算条件は表1に示されています。
表1. 主な計算条件
| 項目 | 設定 |
|---|---|
| 擬ポテンシャル | ウルトラソフト擬ポテンシャル (擬水素Hはノルム保存) |
| 交換相関汎関数 | GGA (PBE) |
| 波動関数のカットオフエネルギー | 30 Rydberg |
| k点サンプリング | 6x12x1 |
| MDアンサンブル | NVT (カノニカルアンサンブル) |
| サーモスタット | Nosé-Hoover |
| 設定温度 | 300 K |
| 時間ステップ | 1 fs |
計算結果と考察#
MDシミュレーションによる構造緩和#
構築した初期構造(図2)を用いて、300 Kの温度設定で第一原理MDシミュレーションを実行しました。図3は、シミュレーション中の系の全エネルギーの時間変化を示しています。シミュレーション開始直後、原子が不安定な初期配置からより安定な配置へと移動するため、全エネルギーは急激に低下します。初期緩和が終わった後、全エネルギーは一定の値に収束せず、ある平均値の周りで揺らいでいるのが見えます。
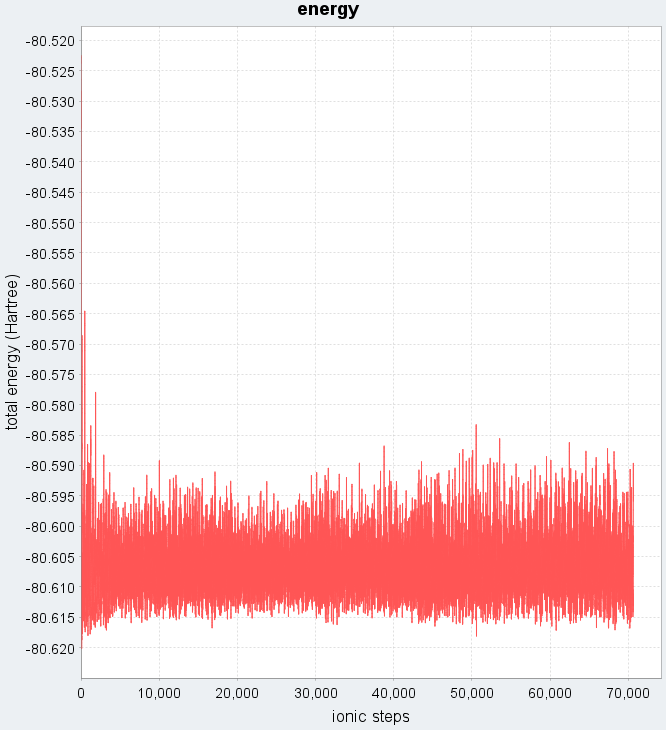
図3. 第一原理MDシミュレーションにおける全エネルギーの時間(ionic step)変化。
N-Nダイマー再構成構造の形成#
図4は、平衡状態に達した後のシミュレーションスナップショット(70,547ステップ目)を示しています。初期構造(図2)では互いに離れていた最表面の窒素原子(N0、緑色で表示)が、MDシミュレーションの過程で自発的に互いに接近し、化学結合を形成していることが明確に確認できます。これが「N-Nダイマー再構成」と呼ばれる構造です。この構造は、c-BN(001)のN終端表面において、エネルギー的に最も安定な構造である [2] と理論的に予測されており、本MDシミュレーションによってその安定な構造が動的に再現されました。文献 [2] によれば、このダイマー再構成による安定化エネルギーは、0 Kにおいて表面の最上層原子あたり1.39 eVと報告されており、これが再構成の強力な駆動力となっています。
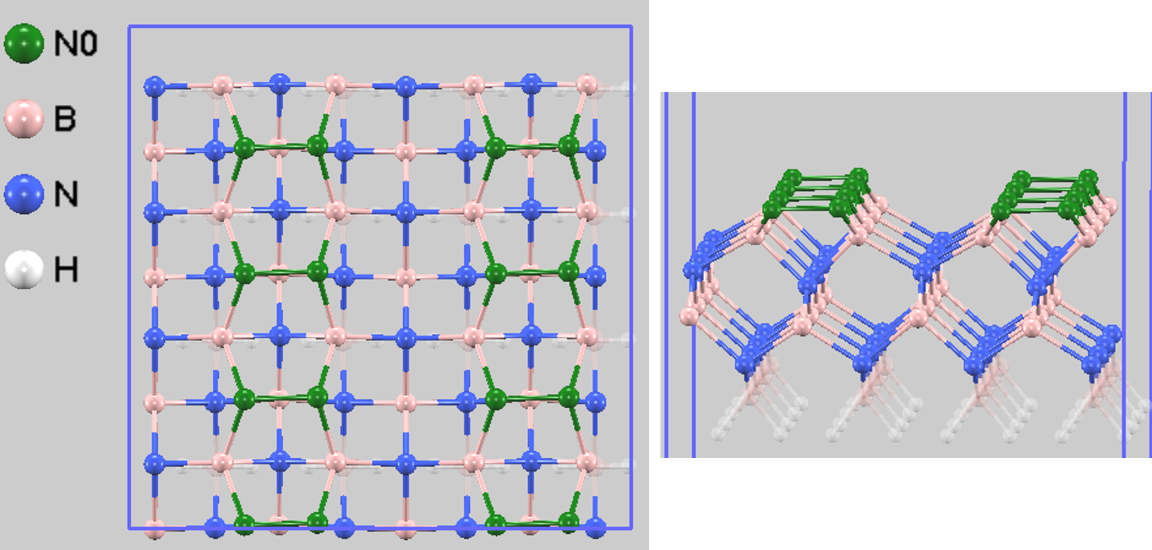
図4. 平衡状態におけるc-BN(001)表面のスナップショット(上面図および斜視図)。最表面のN原子(緑色、N0)が互いに結合し、N-Nダイマーを形成しています。視認性のため、固定層の原子を半透明で表示しています。
N-Nダイマー結合の安定性解析#
次に、形成されたN-Nダイマーが300 Kの温度でどの程度安定であるかを定量的に評価しました。平衡状態に達した50,647から70,647ステップ (約 20 ps)までのMD軌跡データから、ダイマーを形成しているN-N原子間の結合長を毎ステップ記録し、その分布をヒストグラムとしてプロットしました(図5)。
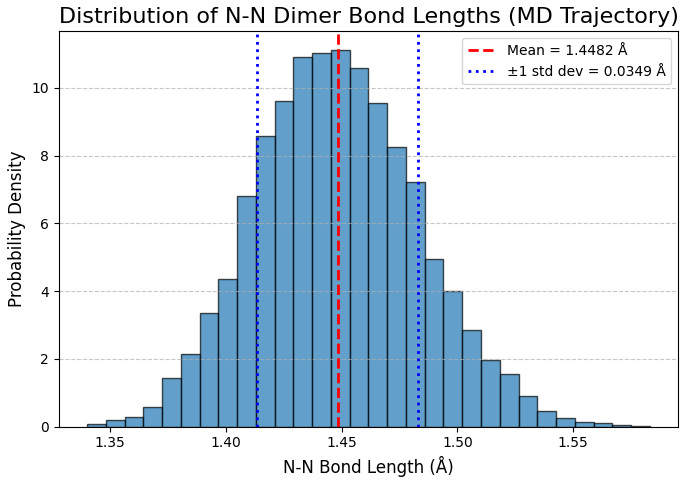
図5. MDシミュレーション軌跡から得られたN-Nダイマー結合長の分布。
図5のヒストグラムは、平均値 1.4482 Å を中心とするシャープな単一のピークを示しています。その標準偏差は 0.0349 Å と想定通り小さく、これはシミュレーション温度(300 K)における熱振動による結合長の変動がごくわずかであることを意味します。この結果は、形成されたN-Nダイマー結合が非常に安定であることを示唆しています。また、本計算(GGA-PBE)で得られた平均結合長 1.4482 Å は、他の文献 [3] の計算値(1.42 Å)とも概ね一致しております。
まとめ#
本解析では、第一原理計算ソフトウェアAdvance/PHASE を用い、c-BN(001)表面のN-Nダイマー再構成について第一原理MDシミュレーションを行いました。 バルクから作成したc-BN(001) 2x1スラブモデルに対し、300 KでのMDシミュレーションを実行しました。その結果、系は安定な平衡状態に達し、最表面のN原子が自発的にN-Nダイマーを形成する再構成過程を動的に観測できました。さらに、平衡状態におけるN-Nダイマー結合長の分布を解析した結果、このダイマー構造が300 Kの熱振動下でも非常に安定であることを示しました。本シミュレーション結果は、c-BN(001)表面の安定構造に関する過去の理論的研究と整合的であり、第一原理MDシミュレーションが表面の動的な再構成プロセスと熱的安定性を解明する上で強力なツールであることを示しています。
本解析の詳細や、研究への適用可能性に関するご相談はこちら
お問い合わせ参考文献#
- E. Knittle, R. M. Wentzcovitch, R. Jeanloz, and M. L. Cohen, "Experimental and theoretical equation of state of cubic boron nitride", Nature 337, 349 (1989).
- J. Yamauchi, M. Tsukada, S. Watanabe, and O. Sugino, "First-principles study on energetics of c-BN (001) reconstructed surfaces", Phys. Rev. B 54, 5586 (1996).
- M. H. Tsai and C. F. Liu, "Reconstruction and electronic structure of the vacancy-free N-and B-terminated c-BN (100) surfaces", Phys. Rev. B 63, 073305 (2001).
関連ページ#
- 第一原理計算ソフトウェア Advance/PHASE
- 解析分野:ナノ・バイオ
- 産業分野:材料・化学