第一原理計算による二次元結晶成長の初期過程解析:h-BNの吸着安定性と配向制御#

グラフェンや六方晶窒化ホウ素(h-BN)など、高品質な二次元材料の製造には、CVD(化学気相成長)法における金属触媒(Ni, Cu等)の利用が不可欠です。特にNi(111)面はh-BNとの格子整合性が極めて高く、欠陥の少ない大面積な単結晶膜が得られることで知られています。本事例では、第一原理計算ソフトウェアAdvance/PHASEを用いて、Ni(111)表面上におけるh-BN単原子層の吸着安定性を評価し、基板がいかにしてh-BNの成長配向(オリエンテーション)を制御しているか、その微視的メカニズムを解明します。
Keywords: 第一原理計算, DFTシミュレーション, h-BN, 二次元材料, Ni(111), 格子整合, 配向制御, 吸着エネルギー, vdW補正, 軌道混成
計算モデルと計算条件#
Ni(111)表面上のh-BN吸着系を模倣するため、13層のNiスラブモデル(両面吸着・対称スラブ)を構築しました。計算精度を確保するため、スラブ中心の3層はバルク位置に固定し、両表面付近の原子(h-BNおよびNi表面5層)の構造最適化を行いました。
格子整合性とエピタキシャル成長の可能性#
高品質な結晶成長において、基板と成長膜の格子定数の一致(格子整合性)は最も重要な因子です。事前にNiバルクとh-BN単層の構造最適化を行い、格子整合性を評価しました。
- Ni バルク格子定数: 3.5729 Å
- これより算出される Ni(111) 表面の原子間距離 Å
- h-BN 格子定数: 2.5177 Å
両者の格子不整合(ミスマッチ)は約 0.3% と極めて小さいことが確認されました [1]。この高い格子整合性は、h-BNがNi基板の原子配列にならって規則正しく成長する「エピタキシャル成長」が可能であることを示唆しています。本解析では、この整合界面を模倣した「1x1 整合モデル」を採用しています。
主な計算条件は表1の通りです。
表1. 主な計算条件
| 項目 | 設定 |
|---|---|
| 計算手法 | 平面波・擬ポテンシャル (DFT) ※ウルトラソフト擬ポテンシャル使用 |
| 交換相関汎関数 | GGA-PBE + D3(BJ) dispersion correction |
| 波動関数のカットオフエネルギー | 30 Rydberg |
| k点サンプリング | 13x13x1 【表面セル:1x1】 |
| スラブ設定 | Ni: 13層 (中心3層固定), h-BN: 両面吸着 |
| スピン分極 | あり (Niの初期スピン分極率: 0.1) |
【解析のポイント】ファンデルワールス力補正の選択
Ni(111)/h-BN系は「弱い化学吸着」と「物理吸着」が競合する難易度の高い系です。通常のPBE汎関数や、単純な分散力補正(PBE+D3 zero-damping)を用いた場合、実験事実とは異なり、表面から3Å以上離れた物理吸着状態が安定化してしまう問題があります [2]。 本解析では、Becke-Johnson (BJ) ダンピングを採用することで、近距離での相互作用を適切に記述し、実験で観測されている化学吸着構造(距離 ~2.2 Å)を高精度に再現することが可能になります。
結果と考察#
1. 吸着構造の安定性と配向制御メカニズム#
h-BNが成長する際、Ni基板に対してどの向き(配向)および位置関係(レジストリ)で安定化するかは、膜の品質を決定づける重要な要素です。複数の吸着配置について計算した結果を表2に示します。
表2. 吸着配置ごとのエネルギーと距離
| 配置名 (N位置-B位置) | 吸着エネルギー (eV / BN pair) |
N-Ni 距離 (Å) | 状態 |
|---|---|---|---|
| N-top_B-fcc | -0.207 | 2.2 | 最安定 (化学吸着) |
| N-top_B-hcp | -0.174 | 2.2 | 準安定 |
| B-top_N-fcc | -0.167 | 3.1 | 物理吸着(エネルギー的に不利) |
| N-fcc_B-hcp | -0.164 | 3.4 | 物理吸着(エネルギー的に不利) |
計算の結果、先行研究の報告 [2, 3] と同様に、N原子がNi原子の直上(Top)、B原子がFCCホローサイトに来る配置(N-top_B-fcc)が最安定であることが判明しました。
ここで注目すべきは、化学吸着状態(最安定)と物理吸着状態のエネルギー差は比較的小さいものの、吸着距離には決定的な乖離(2.2 Å vs 3.1~3.4 Å)が存在する点です。この約1 Åもの構造的な隔たりは、状態間の遷移にポテンシャル障壁が存在することを示唆しています。すなわち、成長初期過程において一度化学吸着距離(~2.2 Å)まで到達したh-BNドメインは、熱的揺らぎによって容易に物理吸着状態へ戻ることはできず、その位置と配向に強く「ロック」されると考えられます。このメカニズムにより、回転ドメインの発生が抑制され、基板全体で向きの揃った高品質な単結晶膜の形成が促進されます。
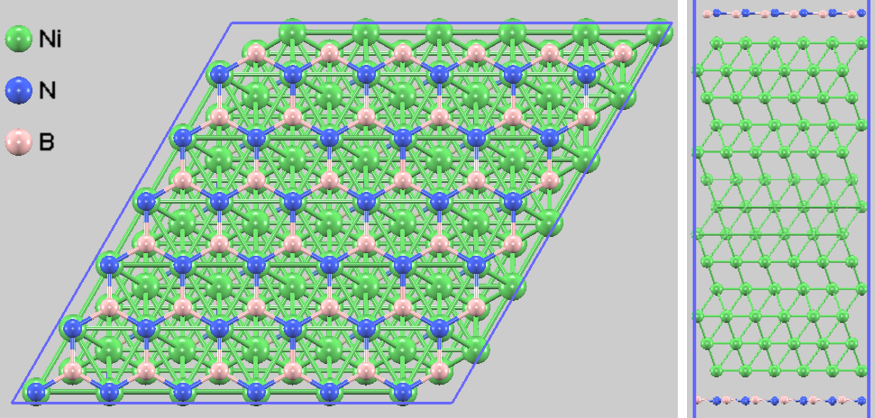
図1. 最安定構造(N-top_B-fcc)の上面図と側面図。視認性のため6x6x1モデルを示しています。
2. 電子状態による結合の可視化#
配向制御の根源にある「化学的な駆動力」を解明するため、「部分電荷密度(Partial Charge Density)」の解析を行いました。
(A) 結合性軌道の形成 (Deep States: -6.0 eV ~ -4.0 eV)#
フェルミ準位よりも深いエネルギー領域(-6.0 eV ~ -4.0 eV)における電荷密度分布を図2に示します。ここでは、N原子の 軌道と、その直下にあるNi原子の 軌道の間にはっきりとつながった電荷の分布(共有結合性)が見て取れます [3]。 この軌道混成(Orbital Hybridization)が、h-BNを基板の特定位置に強く固定し、完全なエピタキシャル成長を実現するための「アンカー」の役割を果たしています [3]。
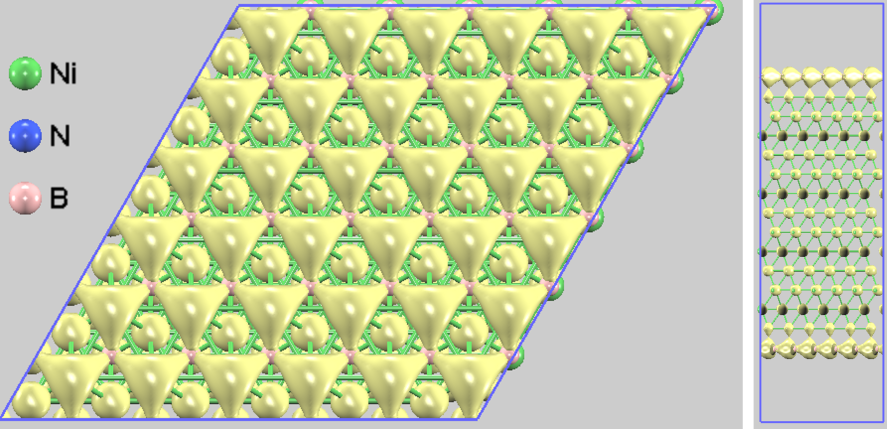
図2. エネルギー範囲 [-6.0, -4.0] eV における部分電荷密度。N原子とNi原子の間に結合性軌道の形成(電荷雲の連結)が明瞭に確認できます。
(B) 界面電子状態 (Shallow States: -0.2 eV ~ 0.0 eV)#
一方、フェルミ準位近傍(-0.2 eV ~ 0.0 eV, VBM付近)の電荷密度分布を図3に示します。ここでは、主にNiのd軌道由来の状態が広がっていますが、界面付近での電荷の再配列も確認できます。この領域の電子状態は、触媒活性や界面でのキャリア輸送特性に影響を与えます。

図3. エネルギー範囲 [-0.2, 0.0] eV における部分電荷密度。フェルミ準位近傍の電子状態を示しています。
まとめ#
本解析では、第一原理計算ソフトウェアAdvance/PHASEを用いてNi(111)表面上のh-BN吸着過程をシミュレーションしました。まず、Ni(111)とh-BNの格子不整合が0.3%と極めて小さいことから、良好なエピタキシャル成長が期待できることを示しました。続いて、適切な分散力補正(PBE+D3 BJ)を用いた構造最適化により、実験事実と整合するN-top_B-fcc構造(吸着距離 ~2.2 Å)が最安定であることを明らかにしました。この特定の配置への強い安定化は、基板による強力な「配向制御」が働いていることを意味します。さらに、部分電荷密度の解析から、N原子の軌道とNiの軌道の強い混成がこの安定化の起源であることを電子論的に実証しました。これらの知見は、Ni基板を用いた高品質な二次元材料の成膜プロセス設計において、重要な指針を与えるものです。
本解析の詳細や、研究への適用可能性に関するご相談はこちら
お問い合わせ参考文献#
- A. Nagashima, N. Tejima, Y. Gamou, T. Kawai, and C. Oshima, "Electronic dispersion relations of monolayer hexagonal boron nitride formed on the Ni(111) surface", Phys. Rev. B 51, 4606 (1995).
- M. N. Huda and L. Kleinman, "h-BN monolayer adsorption on the Ni(111) surface: A density functional study", Phys. Rev. B 74, 075418 (2006).
- G. Grad, P. Blaha, K. Schwarz, W. Auwärter, and T. Greber, "Density functional theory investigation of the geometric and spintronic structure of h-BN/Ni(111) in view of photoemission and STM experiments", Phys. Rev. B 68, 085404 (2003).
関連ページ#
- 第一原理計算ソフトウェア Advance/PHASE
- 解析分野:ナノ・バイオ
- 産業分野:材料・化学