塩害によるCuの初期腐食メカニズムの第一原理解析:表面相図#

銅(Cu)は優れた導電性と加工性を持ち、電子機器や社会インフラに不可欠な材料です。しかし、海洋環境や融雪剤散布環境下においては、塩化物イオン(Cl-)による「塩害」が深刻な問題となります。本事例では、第一原理計算ソフトウェアAdvance/PHASEを用い、腐食反応のトリガーとなる「初期段階」におけるCu表面への酸素(O)と塩素(Cl)の競合吸着挙動を解析しました。実験値を用いた熱力学サイクルによりDFT計算特有のガス分子エネルギー誤差を精密に補正し、酸化(保護皮膜形成)と塩化(腐食進行)のどちらが熱力学的に優先されるかを可視化した「表面相図」を作成しました。
Keywords: 第一原理計算, DFTシミュレーション, 表面相図, Cu腐食, 塩害, CuCl, 吸着エネルギー, 熱力学サイクル
表面相図の理論背景#
第一原理計算は通常、絶対零度(0 K)かつ真空中のエネルギーを出力しますが、実際の腐食現象は有限温度かつ特定のガス分圧下で進行します。このギャップを埋める手法が「第一原理熱力学(Ab initio Thermodynamics)」です。
本手法では、特定の吸着構造の安定性をギブス自由エネルギーの変化 として評価します。
ここで、 と はそれぞれ吸着系と清浄表面のDFT全エネルギー、 は吸着原子数、 はガス種の化学ポテンシャルです。この を温度 や圧力 の関数として変化させることで、環境条件に応じた最安定構造(相)をマップ化したものが「表面相図」となります。
計算モデルと計算条件#
DFT計算は平面波基底・擬ポテンシャル法に基づいています。交換相関汎関数にはGGA-PBEを用い、ファンデルワールス力などの分散力を考慮するためにDFT-D3補正を適用しています。擬ポテンシャルにはウルトラソフト型を使用しました。
本解析では、腐食の最初期段階における反応性を純粋に評価するため、原子間の立体障害や静電反発の影響が少ない低被覆率モデル( ML)を採用しました。これにより、多分子吸着による複雑な相互作用に埋もれることなく、Cu表面が本質的に持っている「酸素および塩素に対する親和性(結合の強さ)」を直接的に比較することが可能です。
表1. 主な計算条件
| 項目 | 設定 |
|---|---|
| 交換相関汎関数 | GGA-PBE + D3 |
| 擬ポテンシャル | ウルトラソフト型 |
| カットオフエネルギー | 35 Rydberg |
| k点サンプリング | スラブモデル:7x7x1 バルク:各相に対して十分に設定 |
| スラブモデル | p(2x2) 単位胞(被覆率 = 0.25 ML), 4層(底面2層固定) |
計算結果と考察#
1. バルク構造の最適化とEOS計算#
まず、Cuの腐食生成物として主要な酸化銅(I) (Cu2O) および塩化銅(I) (CuCl) のバルク構造最適化を行いました。体積を変化させて全エネルギーを計算し、Birch-Murnaghanの状態方程式(EOS)を用いて平衡格子定数と体積弾性率を算出しました。
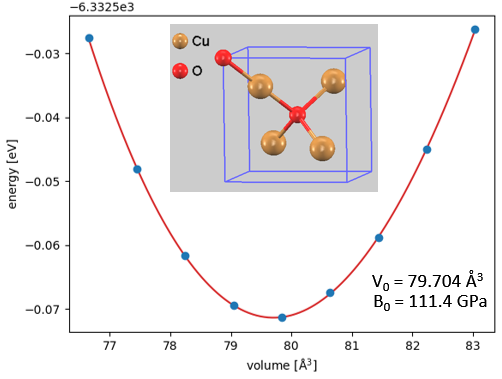
図1. Cu2O (Cuprite構造) のEOS計算結果
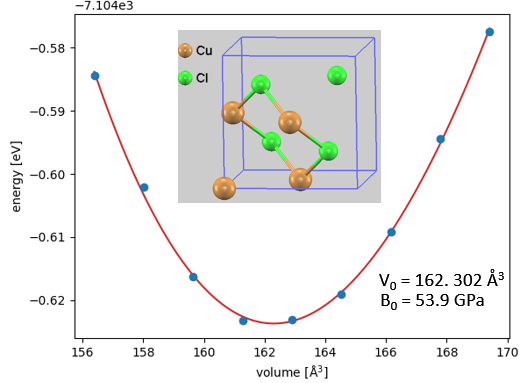
図2. CuCl (Zinc Blende構造) のEOS計算結果
図1と図2で示すように、Cu2OおよびCuClともに、エネルギー曲線は滑らかな放物線を描いており、計算が安定して収束していることが確認できます。算出された格子定数と体積弾性率は実験値と良好に一致しています。
2. Cu(111)表面上の吸着構造解析#
次に、Cu(111)清浄表面に対して酸素(O)原子および塩素(Cl)原子を吸着させたモデルの構造最適化を行いました。
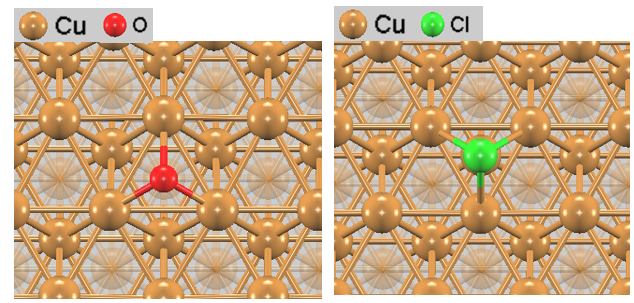
図3. Cu(111)表面上の原子吸着構造。(左)酸素(fccホローサイト)、(右)塩素(hcpホローサイト)
構造探索の結果、O原子はCu(111)面のfccホローサイトに吸着する場合が最も安定であることが確認されました。一方、Cl原子については、fccホローサイトとhcpホローサイトの吸着エネルギー差がわずか数meV程度であり、ほぼ縮退していることがわかりました。これはCl原子が表面上を拡散しやすいことを示唆しています。それぞれの吸着構造を図3で示しています。
3. ガス分子エネルギーの精密補正#
表面相図の作成において、DFT(特にPBE汎関数)におけるO2やCl2ガス分子の結合エネルギー過大評価(Overbinding)は大きな誤差要因となります。これを補正するため、閉殻分子(H2O, HCl)の計算値と実験形成熱を用いた熱力学サイクルを適用しました。
酸素の基準エネルギー ():
塩素の基準エネルギー ():
上記計算式では、298.15 Kにおける形成熱の実験値として、 = -2.506 eV, = -0.957 eVを採用しました [1]。 この補正を適用して吸着エネルギーを再評価した結果、以下の比較が得られました。
- O原子吸着エネルギー: -1.66 eV/atom
- Cl原子吸着エネルギー: -2.09 eV/atom
Clの吸着はOよりも約0.43 eV低く(安定に)なり、塩素が存在する環境下では酸化よりも塩化が優先的に起こりやすいことが示されました。
4. 表面相図(Surface Phase Diagram)と考察#
算出された各エネルギー値に基づき、酸素化学ポテンシャル()と塩素化学ポテンシャル()を軸とした表面相図を作成しました(図4)。
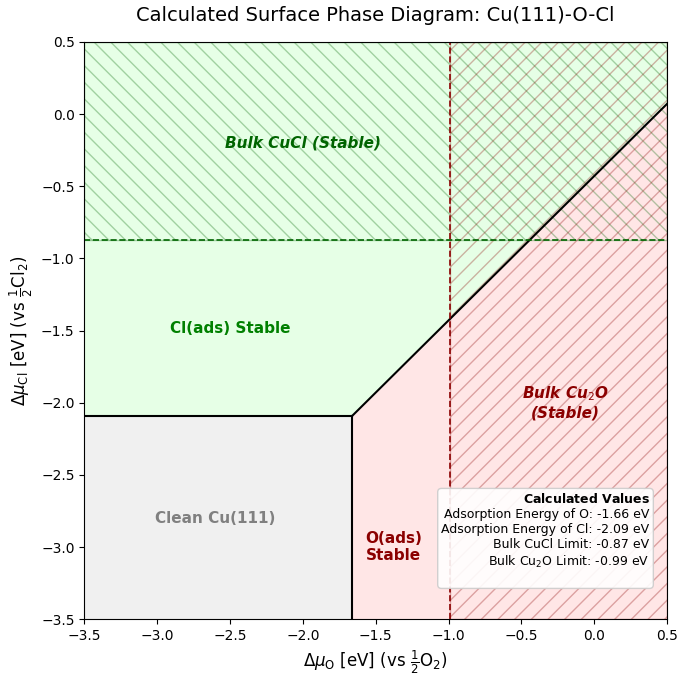
図4. 計算されたCu(111)表面の相図(O-Cl系)
この相図は、横軸が酸素分圧(右ほど酸化環境)、縦軸が塩素分圧(上ほど塩害環境)に対応しており、各環境下でCu表面がどのような状態になるかを示しています。
(1) 吸着エネルギー差に見る「塩害」の駆動力#
相図上の境界線は、以下のエネルギー閾値によって決定されています。
- 酸素(O)吸着の安定限界: eV (赤色領域)
- 塩素(Cl)吸着の安定限界: eV (緑色領域)
ここで注目すべきは、塩素の吸着安定化エネルギー(-2.09 eV)が、酸素(-1.66 eV)よりも約 0.43 eV も深い(安定である)という点です。なお、本計算で得られたCl吸着エネルギー(-2.09 eV)は、分散力補正を含まない先行研究の値(約 -1.83 eV [2] ~ -1.90 eV [3])と比較してより安定な値となっています。これは、DFT-D3補正によってファンデルワールス力が適切に考慮された結果であり、熱力学的に塩素の方が銅表面に対して圧倒的に強い親和性を持つことをより精緻に裏付けています。
図中央の斜めの実線は「酸化と塩化の分岐点(競合ライン)」を表していますが、この線よりも上の領域では、熱力学的に酸素吸着よりも塩素吸着の方が安定となるため、すでに吸着している酸素原子が塩素原子に置換される「駆動力(Driving Force)」が働きます。つまり、わずかな塩素が存在する環境下では、銅本来の保護的な酸化皮膜形成が阻害され、塩化プロセスへと移行するポテンシャルが非常に高いことが、このエネルギー差から理論的に示されました。
(2) バルクCuCl生成と腐食の加速#
図中のハッチング(斜線)領域は、表面吸着だけでなく、バルク状の化合物が成長し始める危険領域を示しています。
- 赤の斜線部 (Bulk Stable): 通常の酸化銅(錆)が生成する領域です。緻密な層であれば母材を保護する役割を果たします。
- 緑の斜線部 (Bulk Stable): eV の領域で、バルクの塩化銅(I)(鉱物名:ナントカイト)が析出します。
特に重要なのが緑色の領域です。ここで生成される CuCl は、水分と反応して加水分解を起こし、塩酸 (HCl) を放出しながら体積膨張を伴って腐食を進行させます(孔食などの自己触媒的腐食)。本計算で示された相図は、塩素ポテンシャルが一定閾値を超えると、表面が単にCl吸着されるだけでなく、この厄介な腐食生成物 CuClへと不可逆的に変化するリスクが高いことを視覚的に示しています。
本計算におけるバルクCuClの生成限界(-0.87 eV)は、文献での先行計算値 (-0.83 eV [4], -0.98 eV [3])と良く一致しており、理論計算としての整合性は取れています。一方で、実験的な形成熱(約 -1.42 eV [3])と比較すると、一般的なGGA汎関数の特性として形成エネルギーが過小評価される傾向にあります。したがって、実際の環境下では、本相図で示された領域よりもさらに低い塩素濃度(低ポテンシャル)からバルクCuClの形成が始まる可能性がありますが、酸化と塩化の相対的な競合関係を理解するための有用な指針が示されています。
また、Cu2Oの生成エネルギーに関しては、GGA-PBEによる過小評価が含まれています。これは遷移金属(Cu)の3d電子に由来する自己相互作用誤差(Self-interaction error)の影響であり、強相関電子系の結合エネルギーを過小評価するGGA汎関数の一般的な傾向によるものです。したがって、図中のCu2O生成ラインは実験値に対して高ポテンシャル側へシフトしていますが、これは計算手法の特性に帰着します。本解析の主眼である「最表面でのOとClの吸着競合」においては、その相対的なエネルギー順位(Clの方が約0.43 eV安定)への影響は小さく、定性的な腐食メカニズムの議論において本結果は十分な妥当性を有しています。
(3) 実環境における腐食進行メカニズム(考察)#
本計算は真空中のモデルですが、Cu表面が熱力学的に酸素よりも塩素に対して強い親和性を持つことが明らかとなりました。実際の湿潤環境では溶媒効果が加わりますが、ここで示された「強い吸着力(Initial Driving Force)」は、実環境においても以下のメカニズム [5] を通じて深刻な腐食を引き起こす「トリガー」になると考えられます。
① 物理的要因:Pilling-Bedworth比(PBR)の不整合
腐食生成膜が母材を保護できるかどうかは、金属が酸化物に変化する際の体積膨張率(PBR)に依存します。一般に、PBRが 1~2 の範囲であれば、被膜は適度な圧縮応力を持ち、緻密で保護性の高い層となります。
本解析のEOS計算(図1)から算出される酸化銅(I)()のPBRは約 1.71 であり、これは母材を保護するのに理想的な値です。通常環境下で銅が耐食性を示すのはこのためです。
一方、計算で予測された塩化銅(I)(、図4の緑色領域)のPBRは約 3.47に達します。これは適正範囲を大きく逸脱しており、生成した層には巨大な内部応力が生じます。この応力を緩和するために被膜は自己崩壊・剥離を起こし、腐食因子(水、Clイオン)の継続的な侵入を許してしまうことが、計算された構造パラメータから強く示唆されました。
Pilling-Bedworth比(PBR)は、金属のモル体積に対する生成化合物のモル体積比として定義され、皮膜の応力状態を予測する指標です。本解析ではEOS計算による平衡体積を用い、次式に基づいて算出しました。
1. 基準パラメータ(Cu原子体積)
FCC-Cuの計算平衡体積より: \( V_{\mathrm{Cu}} \approx 11.68 \, \mathrm{Å}^3/\mathrm{atom} \)
2. 酸化銅(I) (\(\mathrm{Cu}_2\mathrm{O}\)) の評価
単位胞体積 \(V_0=79.704 \, \mathrm{Å}^3\) (\(Z=2\)) より、単位式体積は \(V_{\mathrm{Cu_2O}} = 39.85 \, \mathrm{Å}^3\)。
組成式 \(\mathrm{Cu}_2\mathrm{O}\) より金属消費数 \(n=2\) を適用:
\[ \mathrm{PBR} = \dfrac{39.85}{2 \times 11.68} \approx \mathbf{1.71} \quad (\text{保護性あり: } 1 < R < 2) \]
3. 塩化銅(I) (\(\mathrm{CuCl}\)) の評価
単位胞体積 \(V_0=162.302 \, \mathrm{Å}^3\) (\(Z=4\)) より、単位式体積は \(V_{\mathrm{CuCl}} = 40.58 \, \mathrm{Å}^3\)。
組成式 \(\mathrm{CuCl}\) より金属消費数 \(n=1\) を適用:
\[ \mathrm{PBR} = \dfrac{40.58}{1 \times 11.68} \approx \mathbf{3.47} \quad (\text{保護性なし: } R \gg 2) \]
② 化学的要因:錯イオン形成と自己触媒作用
DFT計算で示された強固な「Cu-Cl結合」は、高濃度の塩化物イオン環境下(水中)においては、逆に腐食を加速させる要因となります。表面に形成されたCuClは、Cl-を取り込んで可溶性の錯イオン([CuCl2]-)を形成し、容易に溶出してしまいます。
さらに、この溶出反応に伴う加水分解によって水素イオン(H+)が発生し、局所的にpHが低下します。酸性化により腐食がさらに進行する「自己触媒的な孔食(Autocatalytic pitting)」のサイクルへと突入します。
まとめ#
Advance/PHASEを用いた第一原理計算により、塩害環境下におけるCuの初期腐食メカニズムを解析しました。熱力学サイクルを用いたガス分子エネルギーの精密補正を行うことで表面相図を作成し、初期吸着過程における腐食の「駆動力」を定量的に評価しました。解析の結果、初期吸着において塩素(Cl)は酸素(O)よりも熱力学的に安定化しやすく(エネルギー差 約0.43 eV)、保護皮膜(酸化銅)の形成よりも塩化反応が優先して進行する傾向が確認されました。この結果は、複雑な環境因子(溶媒効果や多分子吸着など)を考慮する以前の段階で、Cu表面が本質的に「酸素よりも塩素に対して強い親和性」を持つことを明確に示しています。すなわち、この熱力学的に有利な初期吸着が強力なトリガーとなり、その後のPBR不整合による被膜破壊や、錯イオン形成による溶解反応といった深刻な腐食プロセスを誘発することが、原子レベルのシミュレーションから理論的に裏付けられました。
本解析の詳細や、研究への適用可能性に関するご相談はこちら
お問い合わせ参考文献#
- M. W. Chase Jr., NIST-JANAF Thermochemical Tables, 4th Ed., J. Phys. Chem. Ref. Data, Monograph 9 (1998).
- K. Doll and N. M. Harrison, "Chlorine adsorption on the Cu(111) surface", Chem. Phys. Lett. 317, 282 (2000).
- S. Peljhan and A. Kokalj, "Adsorption of Chlorine on Cu(111): A Density-Functional Theory Study", J. Phys. Chem. C 113, 14363 (2009).
- T. V. Pavlova, B. V. Andryushechkin, and G. M. Zhidomirov, "First-Principle Study of Adsorption and Desorption of Chlorine on Cu(111) Surface", J. Phys. Chem. C 120, 2246 (2016).
- G. Kear, B. D. Barker, and F. C. Walsh, "Electrochemical corrosion of unalloyed copper in chloride media—a critical review", Corrosion Science 46, 109 (2004).
関連ページ#
- 第一原理計算ソフトウェア Advance/PHASE
- 解析分野:ナノ・バイオ
- 産業分野:材料・化学