COF-1の電子状態解析と積層構造の安定性評価#

共有結合性有機構造体(Covalent Organic Frameworks, COF)は、その高い多孔性と設計自由度から、ガス吸蔵、触媒、電子デバイスなど幅広い分野で注目されている材料です。特に二次元(2D)COFは、有機分子の層がファンデルワールス力によって積み重なった構造を持ち、その積層の仕方によって物性が大きく変化します。本解析では、代表的な2D COFであるCOF-1を対象とし、第一原理計算ソフトウェアAdvance/PHASEを用いて単層の電子状態を明らかにするとともに、複数の積層構造のエネルギー的な安定性を評価しました。
Keywords: 第一原理計算, DFTシミュレーション, COF-1, 積層安定性, バンド構造, 状態密度, 電荷密度, AA積層, serrated積層
計算モデルと計算条件#
まず、COF-1単層モデルの構造(図1)を最適化し、最も安定な格子定数を決定しました。真空層を20 Åとり、二次元周期的境界条件で計算を行いました。図2に示すように、全エネルギーが最小となる格子定数を求めた結果、最適値として a = 15.20 Å を得ました。これは文献値 [1] の15.11 Åとよく一致しています。
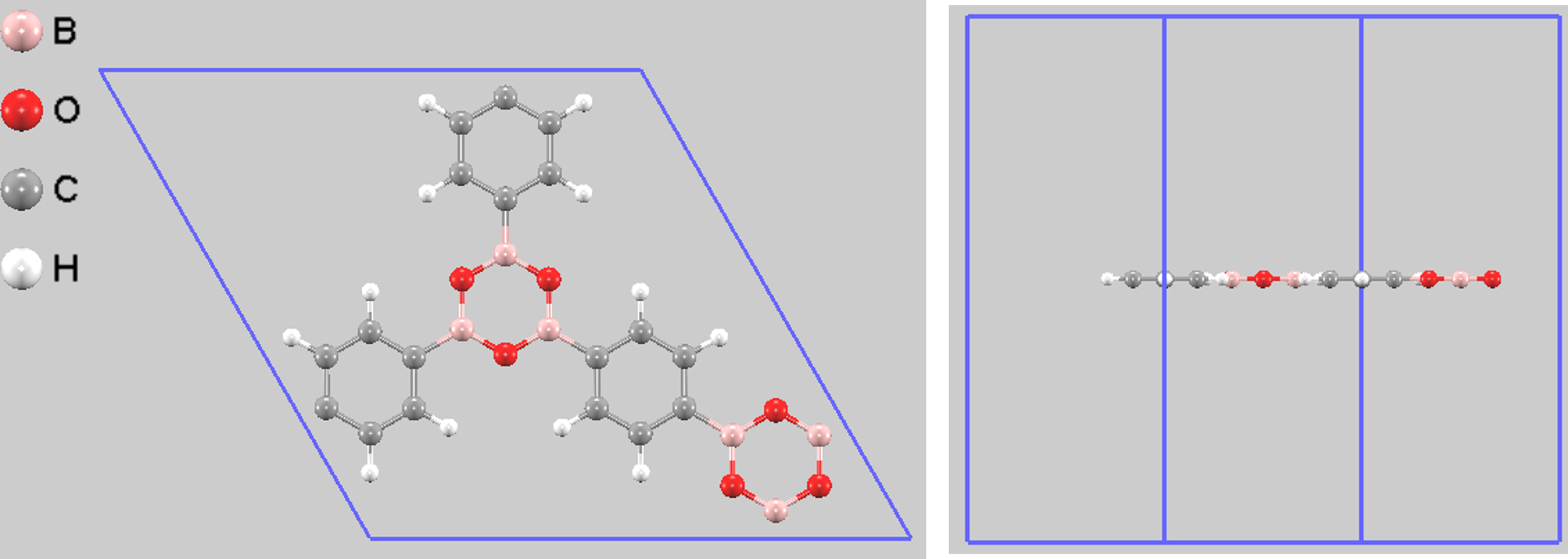
図1. COF-1単層モデルの計算セル(上面図と側面図)。
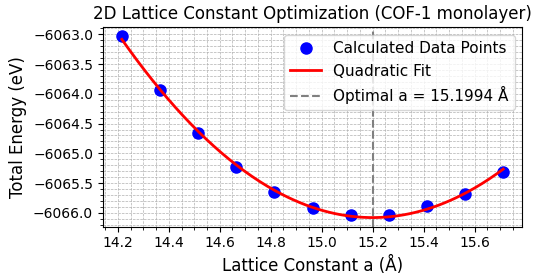
図2. COF-1単層モデルの格子定数最適化。計算から得られた最適格子定数は15.20 Åです。
本解析で用いた主な計算条件を表1に示します。交換相関汎関数にはGGA (PBE)を用いるとともに、層状物質の積層安定性を評価する上で不可欠なファンデルワールス相互作用を考慮するため、DFT-D3による分散力補正を併用しました。なお、ストレステンソルを用いた計算では、カットオフエネルギーをさらに2倍に設定しました。
表1. 計算条件の概要
| 項目 | 設定 |
|---|---|
| 擬ポテンシャル | ウルトラソフト擬ポテンシャル |
| 交換相関汎関数 | GGA(PBE) + DFT-D3 |
| 波動関数のカットオフエネルギー | 25 Rydberg (約340 eV) |
| k点サンプリング | COF平面内: 1x1 (通常SCF計算), 2x2 (エネルギー比較), 層間: 距離に応じて設定 |
計算結果と考察#
COF-1単層の電子状態#
最適化された単層構造について、バンド構造と状態密度(DOS)を計算しました(図3)。計算されたバンドギャップは約3.5 eVでした。これはPBE汎関数に典型的な過小評価を含んだ値であり、実験値はこれより大きいと予想されます。バンド図で特徴的なのは、HOMO(最高被占有分子軌道)に対応する価電子帯の頂上が、エネルギー分散のほとんどない平坦なバンドを形成している点です。これは、HOMOの電子が特定の部位に強く局在していることを示唆しています。
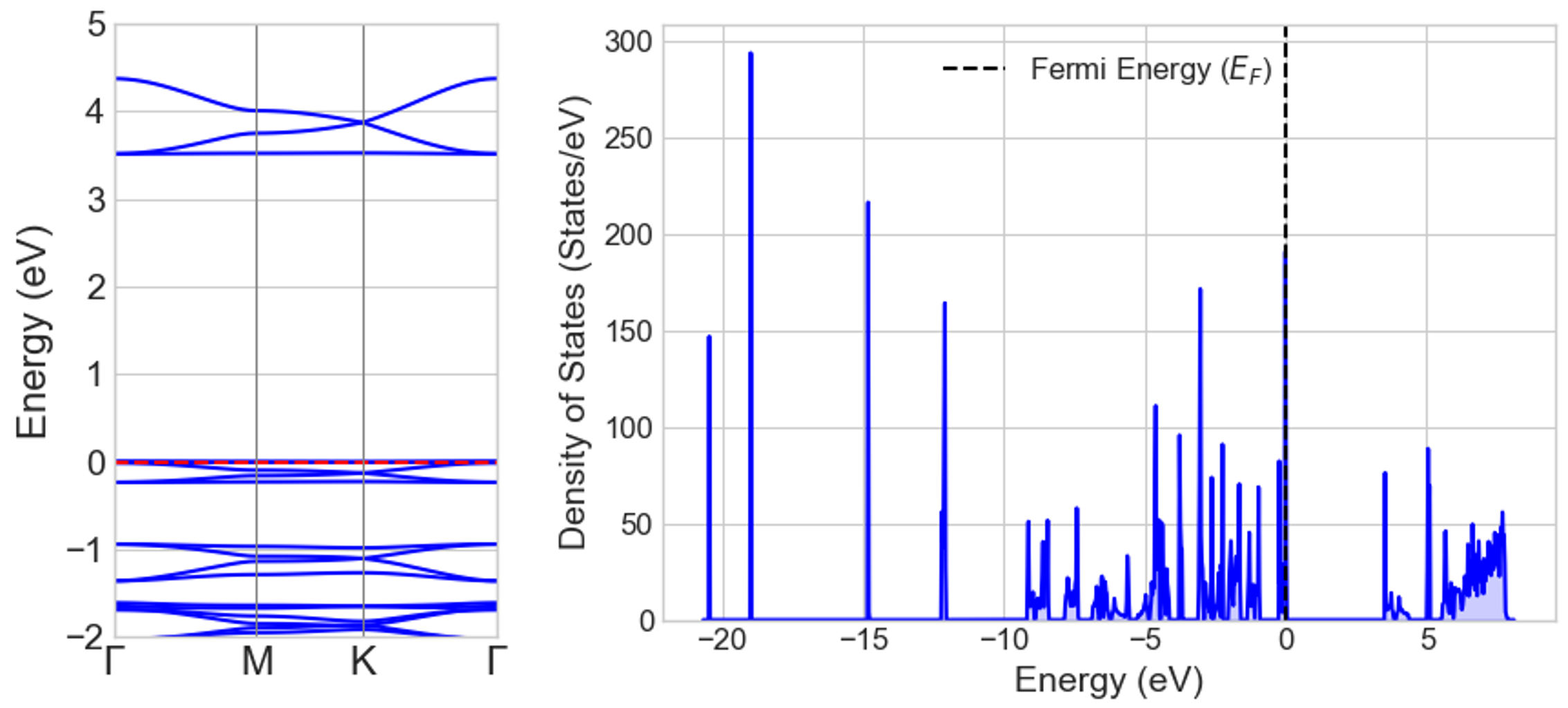
図3. COF-1単層のバンド構造(左)と状態密度(DOS、右)。約3.5 eVのバンドギャップが確認できます。
図4にCOF-1単層の全電荷密度分布を示します。黄色い電子雲は原子間に広がり、C-C結合のような共有結合を明確に示しています。特にB-O結合に注目すると、電子雲が酸素原子側に著しく偏っています。これはホウ素と酸素の大きな電気陰性度の差に起因し、この結合が共有結合でありながらイオン結合性も非常に強い「極性共有結合」であることを示しています。また、フェニル基やボロキシン環の平面上下に広がる電子雲は、材料の電気特性に寄与する非局在化したπ電子の存在を示唆しています。
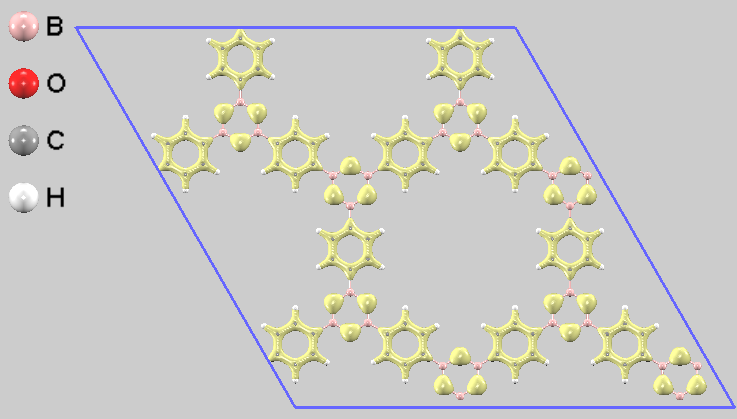
図4. COF-1単層の全電荷密度分布。
電子の局在をより詳細に可視化するため、部分電荷密度を計算しました。その結果、価電子帯の上端から-0.2eVのエネルギー区間(HOMO準位近傍)の電子は主にフェニル基(ベンゼン環)に分布していることが分かります(図5)。一方で伝導帯の下端から+0.2eVのエネルギー区間(LUMO準位近傍)の電子は、ボロキシン環とフェニル基の両方に広がって分布しています(図6)。この電子分布は、COF-1が光励起された際に分子内で電荷の再配置が起こる可能性を示唆しています。
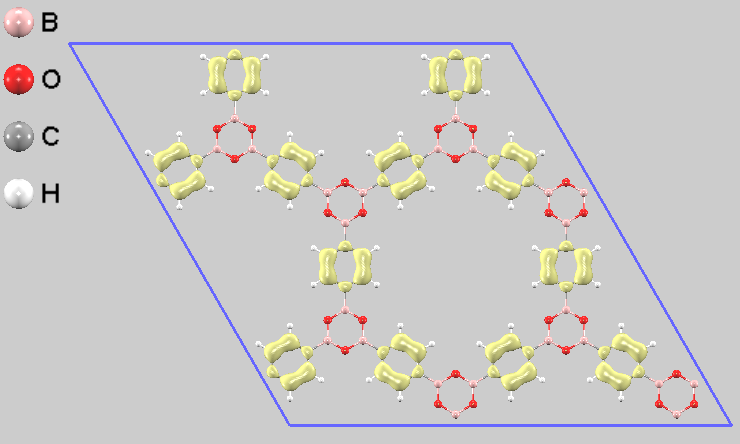
図5. COF-1単層の部分電荷密度分布(HOMO側)。
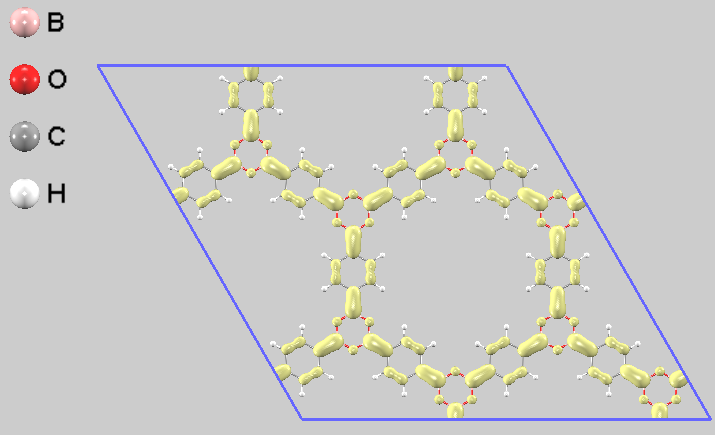
図6. COF-1単層の部分電荷密度分布(LUMO側)。
積層構造の安定性評価#
次に、2D COFの物性を大きく左右する積層構造の安定性を評価しました。ここでは、層が完全に重なり合ったeclipsed (AA stacking) 構造(図7)と、層が面内方向に少しずれたserrated構造(図8)の2種類を比較しました。
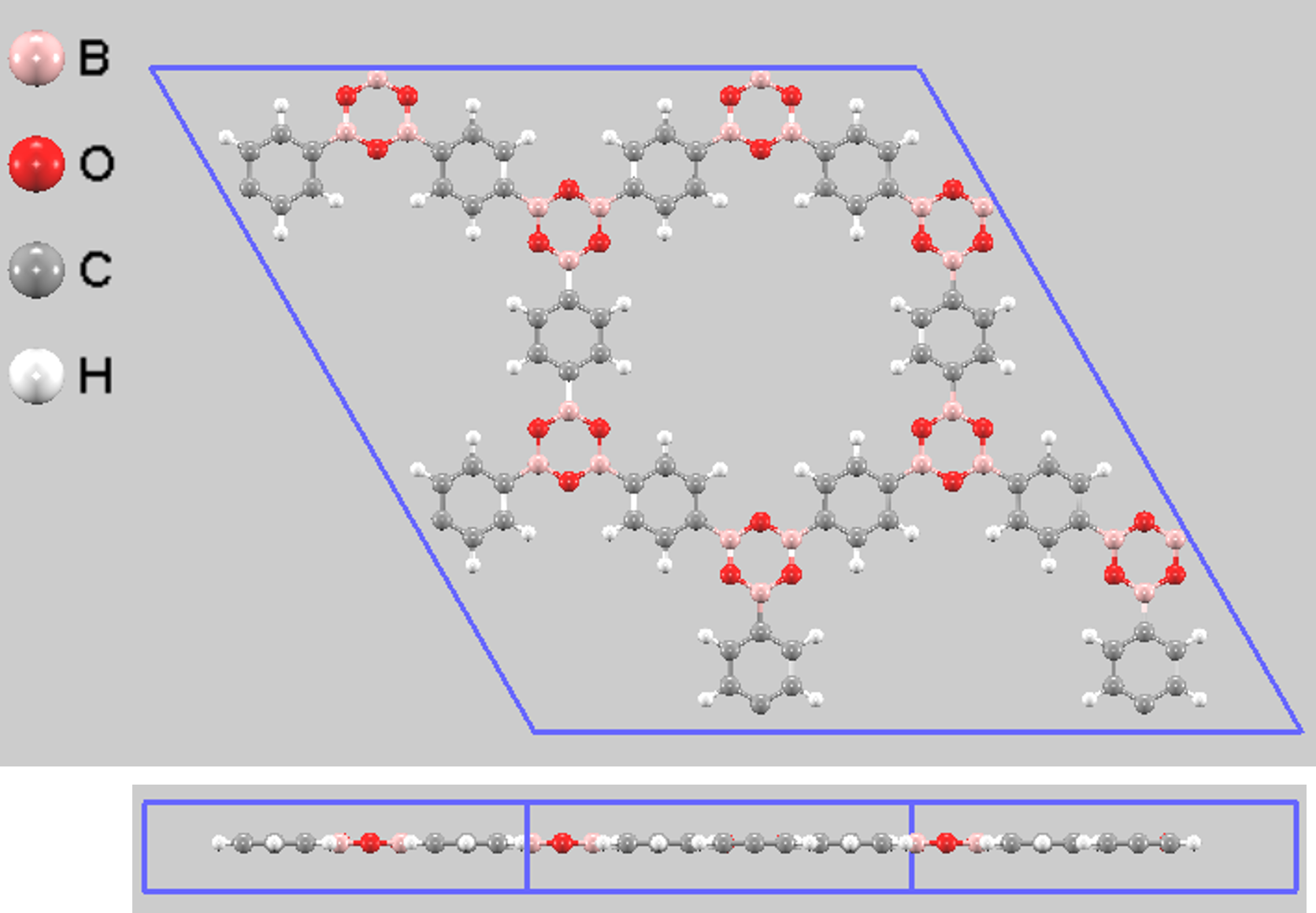
図7. Eclipsed (AA) 積層構造モデル(上面図と側面図)。視認性向上のため2x2x1スーパーセルを示しています。
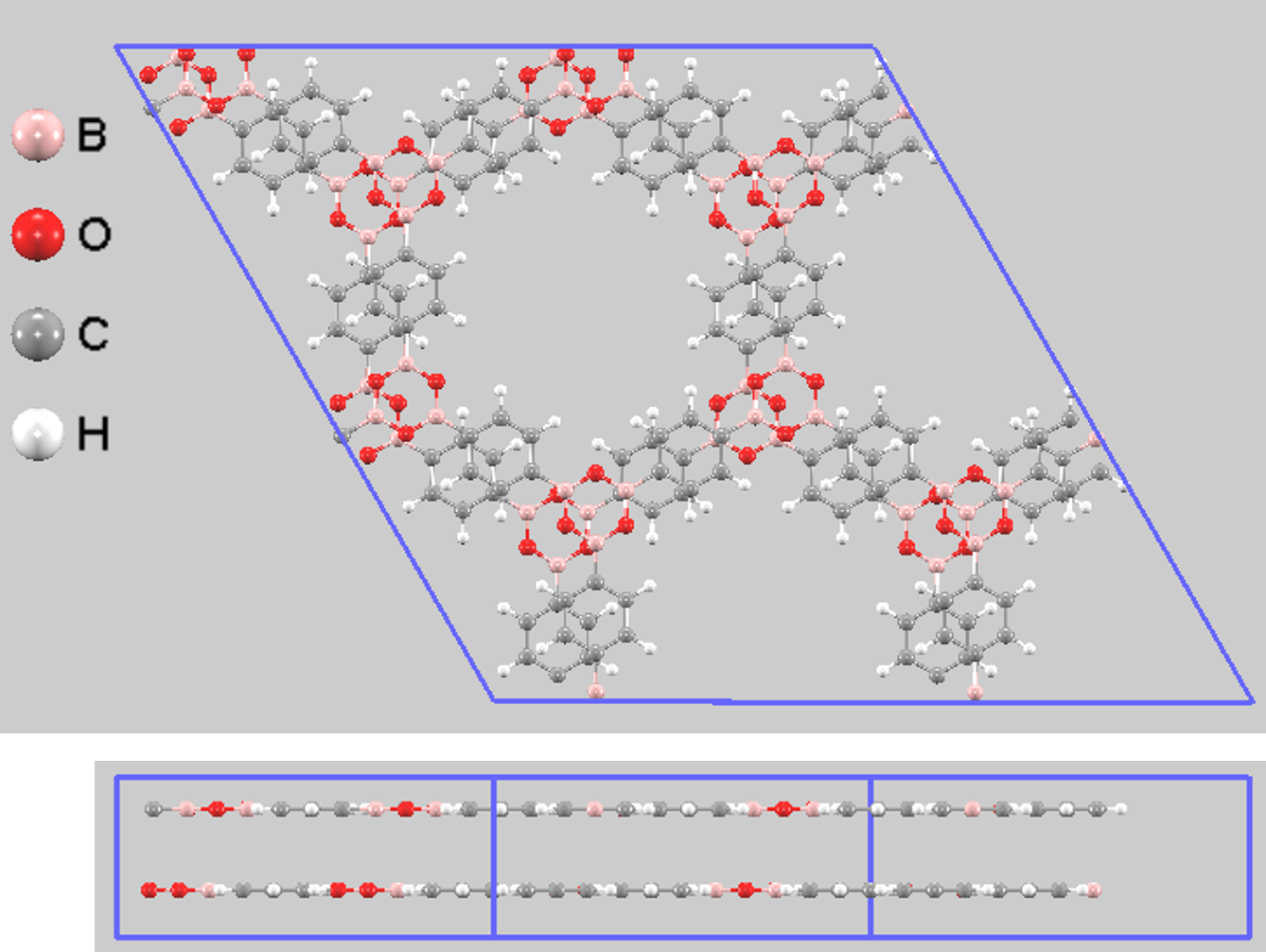
図8. Serrated積層構造モデル(上面図と側面図)。視認性向上のため2x2x1スーパーセルを示しています。
各モデルについて格子定数最適化・構造最適化を行った結果と、両者のエネルギー差を表2にまとめます。計算の結果、serrated構造はeclipsed構造に比べて 40.73 kJ/mol (1層あたり0.422 eV/cell)安定であることが明らかになりました。この結果は、COF-1が単純な重なり構造よりも、層が互いにずれた配置を好むことを示しており、π電子雲の反発を避け、より有利なπ-π相互作用(πスタッキング)を形成するためだと考えられます [2, 3]。このエネルギー差は、複数の文献値とも整合的な値です。
また、serrated構造では層間距離が短くなっており(3.23 Å)、より密なパッキングを形成していることがわかります。
表2. 各積層構造の最適化結果とエネルギー差
| 構造 | 面内シフト量 (Å) | 層間距離 (Å) | 相対エネルギー (kJ/mol) |
|---|---|---|---|
| Eclipsed (AA) | 0 | 3.50 | 40.73 |
| Serrated | 1.75 | 3.23 | 0 (基準) |
なお、文献 [2] では、serrated構造と非常にエネルギーが近いincline構造の存在も示唆されており、実際のCOF-1材料 [4] 中ではこれらの構造が混在している可能性も考えられます。
まとめ#
本解析では、第一原理計算ソフトウェアAdvance/PHASEを用い、COF-1の電子状態解析と積層構造の安定性評価を行いました。単層モデルの計算からは、約3.5 eVのバンドギャップや、HOMOとLUMOに由来する電子の局在性が明らかになりました。積層モデルの安定性評価では、層がずれたserrated構造が、完全に重なったeclipsed構造よりも約40.7 kJ/molも安定であることを定量的に示しました。この安定化は、より有利なπスタッキング相互作用に起因するものと考えられます。これらの結果は、COFの物性発現メカニズムの理解や、新規材料設計への指針を与えるものであり、第一原理計算が複雑なナノ材料の解析に強力なツールであることを示しています。
本解析の詳細や、研究への適用可能性に関するご相談はこちら
お問い合わせ参考文献#
- H. Zhao, Y. Guan, H. Guo, R. Du, and C. Yan, "Hydrogen storage capacity on Li-decorated covalent organic framework-1: A first-principles study", Materials Research Express 7, 035506 (2020).
- K. S. Rawat, S. Borgmans, T. Braeckevelt, C. V. Stevens, P. Van Der Voort, and V. Van Speybroeck, "How the layer alignment in two-dimensional nanoporous covalent organic frameworks impacts its electronic properties", ACS Applied Nano Materials 5, 14377 (2022).
- C. Winkler, T. Kamencek, and E. Zojer, "Understanding the origin of serrated stacking motifs in planar two-dimensional covalent organic frameworks", Nanoscale 13, 9339 (2021).
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger, and O. M. Yaghi, "Porous, crystalline, covalent organic frameworks", Science 310, 1166 (2005).
関連ページ#
- 第一原理計算ソフトウェア Advance/PHASE
- 解析分野:ナノ・バイオ
- 産業分野:材料・化学