色素増感太陽電池(DSSC)における色素分子とTiO2のバンドアライメント評価#

色素増感太陽電池(DSSC)の性能向上には、太陽光を効率よく吸収する「色素」と、電子を輸送する「酸化チタン(TiO2)」の界面におけるエネルギー準位の最適な配置(バンドアライメント)が不可欠です。本事例では、第一原理計算ソフトウェアAdvance/PHASEの高度な固体・表面計算機能と、オープンソースの量子化学計算ソフトNWChemを連携させ、計算コストを抑えつつ高い物理的整合性を持つDSSC材料のインシリコ・スクリーニングフローを紹介します。
Keywords: 第一原理計算 (DFT), NWChem, 色素増感太陽電池 (DSSC), バンドアライメント, TD-DFT, HSE06汎関数
理論背景#
DSSCの計算化学的アプローチにおいて、系を「分子(色素)」と「固体表面(TiO2)」に分割し、それぞれに適した手法を用いて絶対エネルギー準位を評価する手法は、大規模な計算を避けるための有効な戦略です [1, 2]。本解析では、固体側の正確な絶対表面電位をAdvance/PHASE(HSE06汎関数 + ESM法)で、分子側のエネルギー準位と光吸収特性をハイブリッド汎関数を用いたNWChem(TD-DFT)で算出します。
DSSC色素のスクリーニングにおける2つの必須条件
優れた色素材料を設計するためには、以下の「光吸収」と「電子移動」の要件を同時に満たす必要があります。
- 光吸収特性(TD-DFT法で評価): 太陽光エネルギーを無駄なく利用するため、可視光から近赤外領域にかけて強い光吸収(高い振動子強度)を示すこと。
- エネルギー準位の整合性:
(1) 電子注入: 光励起された状態のLUMO(LUMO*)がTiO2の伝導帯下端(CBM)より高く、電子注入の駆動力が確保されていること。このLUMO*は、基底状態の軌道エネルギー(HOMO)にTD-DFTで算出した光学ギャップ(励起エネルギー)を加算することで、励起子効果を含めた物理的に妥当な値として評価します。
(2) 色素再生: 色素の最高被占軌道(HOMO)が電解液の酸化還元電位より低く、失った電子を速やかに補填できること。
Advance/PHASEによるTiO2表面の高精度解析#
1. HSE06汎関数によるバルク・バンドギャップの補正#
一般的なGGA汎関数(PBEなど)は、半導体のバンドギャップを過小評価する傾向があります。Advance/PHASEを用いてルチル型TiO2バルクの状態密度(DOS)を計算した結果を図1に示します。PBE計算値(オレンジ)の1.63 eVに対し、ハイブリッド汎関数であるHSE06計算値(青)ではバンドギャップが大きく開き、実験値(3.0 eV)に近い3.06 eVを与えることが確認されました。
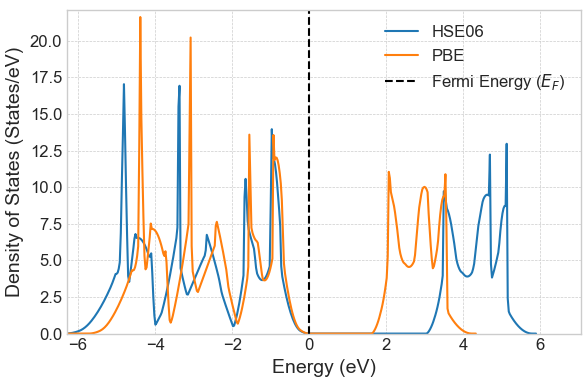
図1. ルチル型TiO2バルクのDOS計算結果。
2. ESM法による絶対表面電位の算出#
TiO2のバンドエッジ(VBM, CBM)を真空準位基準の絶対値で評価するため、熱力学的に最も安定なルチル型TiO2 (110)面のスラブモデルを作成しました。この際、計算コストの低減のため、中心層を固定して両表面のみを緩和させる手法を採用しています(図2)。
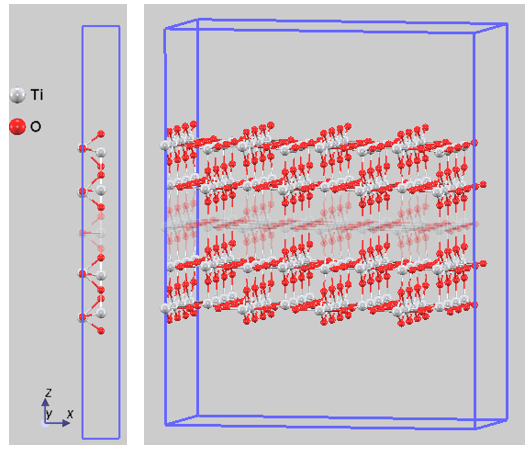
図2. ルチル型TiO2(110)面のスラブの構造モデル(最適化済)。左:計算セル、右:視認性のため4x4x1スーパーセル表示。中間の原子層(半透明表示)はバルク状態を保つよう固定されています。
作成したスラブモデルに対し、仕事関数やイオン化ポテンシャルを高精度に算出できるESM(Effective Screening Medium)法 [3] を適用しました(図3)。半導体表面であるルチル型TiO2(110)に対して、このESM法によって得られた 7.77 eV というイオン化ポテンシャルは、Kangらによる高度なGW法を用いた理論計算値(7.8 eV)[4] と非常に良好な一致を示しており、ESM法を用いた解析精度の高さが示されました。これにより、TiO2の伝導帯下端(CBM)は -4.71 eV (= -7.77 + 3.06)と正確に決定されました。
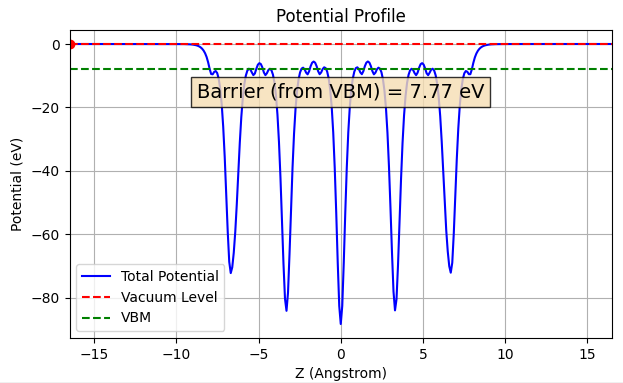
図3. ルチル型TiO2(110)面の垂直方向(z方向)における面内平均ポテンシャルのプロファイル。真空準位(Vacuum Level)と価電子帯上端(VBM)のエネルギー差(イオン化ポテンシャル)は 7.77 eVと算出されました。
NWChemによる色素分子の光吸収特性 (TD-DFT)#
分子側のエネルギー準位(HOMO, LUMO)と光吸収特性解析では、NWChem [5] の多様な量子化学計算機能を活用しています。特にNWChemは、ハイブリッド汎関数を用いたTD-DFT計算が可能であり、色素分子の励起エネルギーや吸収スペクトルを高精度に評価するのに適しています。本解析では、B3LYP/6-311+G**レベルでアリザリンおよびクマリン343の構造最適化(図4)を実施し、その励起状態を算出しました(図5)。
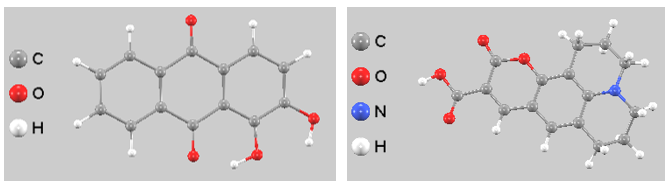
図4. 計算に用いた色素分子の最適化構造。左:アリザリン(Alizarin)、右:クマリン343(Coumarin 343)。
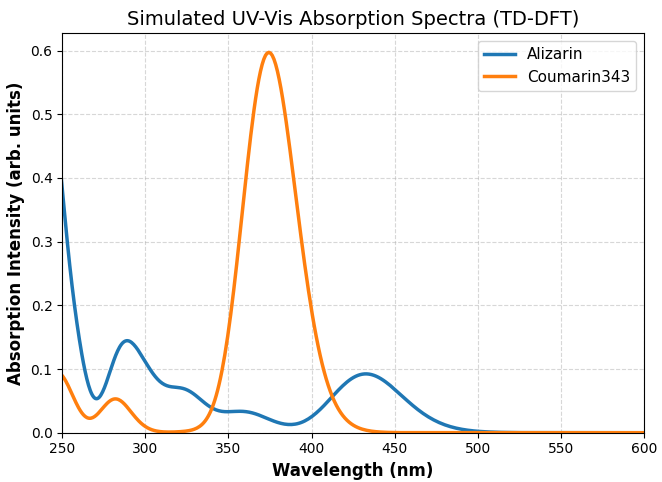
図5. TD-DFT (B3LYP/6-311+G**) 計算で得られた色素分子の光吸収スペクトル(ガウス関数による広がり付与)。
【考察】
図5より、クマリン343は可視光領域(約 374 nm)に強い吸収を示し、典型的なDSSC有機色素の特性を表しています。この計算結果は、ガス相における既存の計算化学の文献値とよく整合しています [6]。そして、これと同様の計算設定を用いてアリザリンの光吸収特性を解析したところ、約 433 nm に励起状態を持つものの、その振動子強度は小さく、主ピークが紫外領域(約 243 nm)に位置する結果となりました。
界面のエネルギー・バンドアライメント評価#
Advance/PHASE(TiO2の絶対電位)とNWChem(色素の励起状態)の結果を統合し、真空準位を基準としたエネルギー・バンドアライメントを作成しました(図6)。DSSCにおける電子注入は光励起状態から起こるため、単なるLUMOの軌道エネルギーではなく、基底状態のHOMOにTD-DFTの光学ギャップ(第1励起エネルギー)を加算した LUMO* をプロットすることで、より物理的実態に即した評価を行っています。
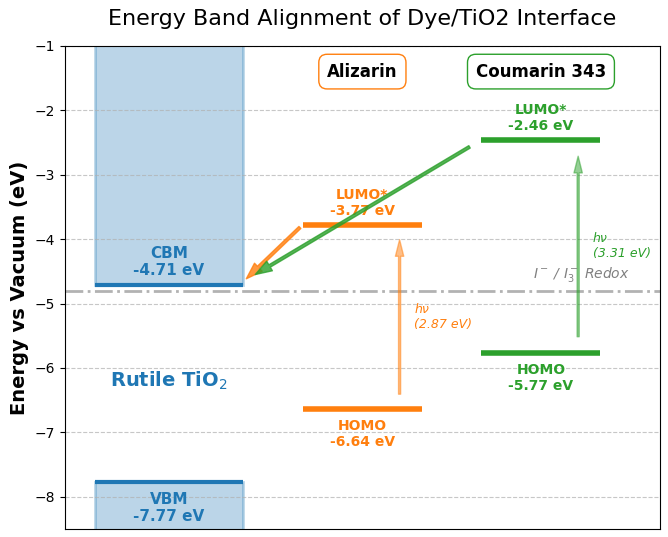
図6. 色素分子(Alizarin, Coumarin 343)とルチル型TiO2 (110)面のエネルギーバンドアライメント図。LUMO*は光励起状態のエネルギーレベルを示します。
【考察】
図6より、どちらの色素分子においても、光励起状態であるLUMO*準位(アリザリン: -3.77 eV, クマリン343: -2.46 eV)が、TiO2の伝導帯下端(CBM: -4.71 eV)よりも十分に高い(浅い)位置にあることが示されています。このエネルギー的な落差により、励起子効果を考慮した上でも光励起された電子は色素からTiO2へ向かって自発的に注入されることが裏付けられました。さらに、色素のHOMO準位は、図中のI-/I3-電解液の酸化還元電位(-4.8 eV)よりも十分に深い位置にあり、電子を失った色素分子が電解液からスムーズに電子を受け取り再生可能であることを示しています。このように、HOMOとLUMO*を組み合わせて図示することで、TD-DFTをスクリーニングに組み込む理論的妥当性が明確になります。
発展的考察:インシリコ・スクリーニングと実験的検証の相補的協働#
本事例では、色素分子およびTiO2表面のいずれも真空準位を基準とした「真空近似モデル」を採用しています。実際のDSSCデバイスは電解液(溶媒)環境下にありますが、計算材料科学、特にマテリアルズ・インフォマティクス(MI)の実務において、この近似は合理的な戦略的アプローチとして位置づけられます。
- 相対的序列(トレンド)の保存: 溶媒効果の導入はエネルギー準位や吸収波長の絶対値をシフトさせますが、類似骨格を持つ分子群におけるパフォーマンスの相対的な序列は、多くの場合、真空中の計算においても高度に保存されます。このため、有力候補を抽出する「トレンド解析」において真空近似は有効です。
- 階層的スクリーニングの最適化: 真空近似による計算は、環境依存のパラメータを排除できるため有効かつ低コストです。多数の候補化合物から、熱力学的条件を明らかに満たさない分子を排除する「Tier 1(一次)スクリーニング」として、実用的なフィルタリング手法となります。
【計算スクリーニングから実験的検証への連携】
現実の材料開発において、複雑な固液界面や溶媒和構造のすべてを考慮した高精度シミュレーションを全候補に適用することは、計算コストおよび計算設定の複雑性の観点から現実的ではありません。材料探索の成功確率を最大化するワークフローは、本事例に示した「物理的根拠に基づく低コストなインシリコ・スクリーニング」によって有望な候補を迅速に絞り込み、得られた知見を速やかに合成・デバイス評価へとフィードバックすることです。
なお、本解析は真空近似モデルに基づく相対的な評価であり、実際の開放電圧(Voc: CBMと電解液の酸化還元電位のエネルギー差)の評価や界面での複雑な電荷移動を精密に議論するには、固液界面モデルや実験データとの詳細な照合、溶媒効果の導入といった更なる深掘りが重要となります。このような計算機と実験の協働が、効率的な材料開発において重要となります。
まとめ#
本事例では、第一原理計算ソフトウェアAdvance/PHASEによる高精度な表面電位評価と、量子化学計算パッケージNWChemのハイブリッド汎関数を用いたTD-DFT解析を組み合わせることで、色素増感太陽電池の信頼性の高いバンドアライメント評価を実現しました。この低コストな分割評価ワークフローは、新しい色素材料のハイスループット・スクリーニングにおいて有用な手法となります。
本解析の詳細や、研究への適用可能性に関するご相談はこちら
お問い合わせ参考文献#
- M. Pastore, E. Mosconi, F. De Angelis, M. Grätzel, "A Computational Investigation of Organic Dyes for Dye-Sensitized Solar Cells: Benchmark, Strategies, and Open Issues", J. Phys. Chem. C 114, 7205 (2010).
- F. De Angelis, "Modeling Materials and Processes in Hybrid/Organic Photovoltaics: From Dye-Sensitized to Perovskite Solar Cells", Acc. Chem. Res. 47, 3349 (2014).
- M. Otani and O. Sugino, "First-principles calculations of charged surfaces and interfaces: A plane-wave nonrepeated slab approach," Phys. Rev. B 73, 115407 (2006).
- W. Kang and M. S. Hybertsen, "Quasiparticle and optical properties of rutile and anatase TiO2", Phys. Rev. B 82, 085203 (2010).
- E. Aprà et al., "NWChem: Past, present, and future", J. Chem. Phys. 152, 184102 (2020).
- W. Wu, Z. Cao, and Y. Zhao, "Theoretical studies on absorption, emission, and resonance Raman spectra of Coumarin 343 isomers", J. Chem. Phys. 136, 114305 (2012).
関連ページ#
- 第一原理計算ソフトウェア Advance/PHASE
- 解析分野:ナノ・バイオ
- 産業分野:材料・化学