表面Pourbaix図を用いたPt(111)酸化被膜形成の第一原理解析:真空モデル評価と溶媒寄与#

燃料電池や水電解装置の電極触媒において、白金(Pt)は最も基本的かつ重要な材料です。実際の電気化学環境下では、電位の上昇に伴いPt表面が酸化され、その触媒活性が変化します。本解析では、第一原理計算ソフトウェアAdvance/PHASEを用い、Pt(111)表面における酸素(O)および水酸基(OH)の吸着状態を評価しました。本事例では、計算コストを抑えた最も基礎的なアプローチとして真空(Dry)環境モデルを採用し、計算水素電極 (CHE) モデルを用いて表面プルベー図(Surface Pourbaix Diagram)を作成しました。得られた結果と実験事実との差異から、電極表面における「水分子(溶媒)の役割」について考察します。
Keywords: 第一原理計算 (DFT), 表面Pourbaix図, Pt(111), OER, ORR, CHEモデル, 溶媒和効果
理論的背景と計算手法#
吸着エネルギーの定義#
本解析では、水分子と水素分子を基準とし、以下の表面反応式を想定して吸着エネルギーを算出しました (*は表面上の吸着サイトを表します)。
酸素吸着:
OH吸着:
これに基づき、吸着種1個(原子または分子)あたりの吸着エネルギー は以下の式で定義されます。ここで は単位格子内の吸着種の数です。
※本定義では、 が負であるほど(あるいは小さいほど)、吸着状態が安定であることを示します。
CHEモデルによる自由エネルギー計算#
Nørskovらが提唱した計算水素電極 (CHE) モデル [1] を用い、電圧印加下における表面の安定性を評価しました。電位 (vs RHE) における吸着種1個あたりのギブス自由エネルギー は次式で表されます。
ここで、 はDFT計算による吸着エネルギー、 と はそれぞれ零点振動およびエントロピー補正項です。また、 は吸着種1個の生成反応に伴い移動する電子の数(化学量論数)です。具体的には、OH吸着に対し 、O吸着に対し の定数となります。
本解析では、Rossmeislら [2] の手法に準拠し、以下の標準的な熱力学補正項 を採用しました。なお、これらの値は気相分子の実験値およびCu(111)表面上の吸着種の振動計算値に基づき算出されたものであり、同分野のトレンド解析において広く用いられている値です。
- OH吸着: ( 基準)
- O吸着: ( 基準)
表面Pourbaix図の構築と解釈#
- 構築手順(Construction): 検討する全ての吸着構造(Clean, OH, Oなど)について、自由エネルギー を電位 に対してプロットします。各相は、移動電子数 に対する傾きと切片を持つ一次関数として描画されます。
- 解釈(Interpretation): ある電位 において、最もエネルギーが低い(グラフの最下部に位置する)相が、その環境下で熱力学的に実現する安定状態です。異なる相の直線が交差する点は、表面状態が切り替わる「相転移点(酸化還元電位)」を意味します。
計算モデル#
計算にはPt(111)表面の p(2x2) スーパーセル(3層スラブモデル)を用いました。表面上のfcc hollowサイトやtopサイトなどに対し、O原子またはOH基を配置し、被覆率(Coverage)を変化させて(0.25 ML ~ 1.00 ML)構造最適化を行いました。
表1. 計算条件の概要
| 項目 | 設定 |
|---|---|
| 擬ポテンシャル | ウルトラソフト擬ポテンシャル (H: ノルム保存) |
| 交換相関汎関数 | GGA (RPBE + D3補正) |
| 波動関数のカットオフエネルギー | 25 Rydberg |
| k点サンプリング | 6x6x1 |
| 溶媒モデル | なし(真空モデル) |
計算結果#
1. 構造最適化結果#
構造最適化の結果、各吸着種の安定サイトは以下のようになりました。これらの結果は文献 [3] と一致しています。
- 酸素(O): 全ての被覆率において fcc hollowサイト が最安定。
- 水酸基(OH): 全ての被覆率において topサイト が最安定。
図1に被覆率1.0 MLでの最適化構造を示します。OH基(図1右)は高被覆率において水素結合ネットワークを形成し、特定の配向を持つことが確認されました。
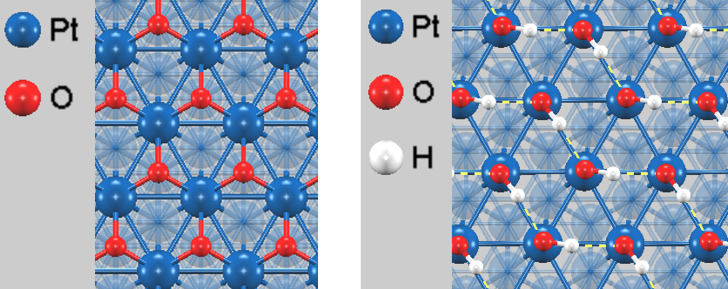
図1. 被覆率1.0 MLで最適化された吸着構造。(左) O吸着 @ fccサイト, (右) OH吸着 @ topサイト。破線は水素結合を示しています。
2. 表面Pourbaix図(真空モデル)#
算出された自由エネルギーに基づき、各電位で最も安定な表面状態をプロットした表面Pourbaix図を図2に示します。
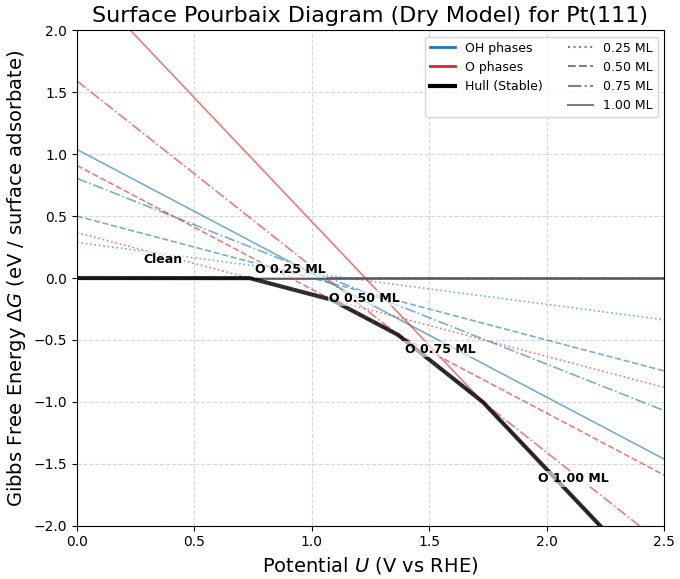
図2. Pt(111)表面の表面Pourbaix図(標準真空モデル)。太い黒線がその電位における最安定相を示す。
図2の結果から、以下の相転移挙動が得られました。
- 0 V ~ 0.73 V付近: クリーンなPt表面が安定。
- 0.73 V以上: O吸着相(0.25 ML)が出現し、電位上昇とともにO被覆率が増加(0.25 ML → 0.50 ML → 1.00 ML)。
- OH相の不在: 実験で観測される「OH吸着相」は、どの電位領域でも最安定相として現れませんでした(青色のOH線が赤色のO線より常に上にあるため)。
※Pt電極の酸化劣化(Pt-O形成)の開始電位について、真空モデルによる計算の結果、0.73 V (vs RHE)でO吸着が始まると予測されました。これは、詳細な溶媒モデルを用いた計算値(0.78 V) と0.05 V以内の差で一致しており、O原子に対する溶媒効果が限定的であることを示しています。
考察:なぜOH相が現れないのか?#
本解析結果では、実験的に知られている「Clean → OH → O(酸化)」という順序 [4, 5]ではなく、OH相を経由せず直接O相(酸化被膜)へ遷移する結果となりました。これは計算自体の問題ではなく、「真空環境」を仮定した物理モデルの特性を反映しています。
溶媒和効果(Solvation Effect)の重要性#
先行研究 [1, 2, 6] によれば、OH基とO原子では水分子との相互作用の強さが大きく異なります。
- OH基: 極性を持ち、周囲の水分子と水素結合を形成しやすいため、水が存在することで 約 0.3 ~ 0.5 eV 安定化します。
- O原子: 水分子との相互作用は比較的弱く、安定化効果は限定的です。
本解析は真空モデルであるため、OH基に対するこの「水による安定化エネルギー」が含まれていません。その結果、相対的にO吸着の方が有利(エネルギーが低い)と評価され、OH相が図から消失したと考えられます。
より現実の環境に近い解析を行うためには、以下の手法が有効です。
- 静的な水分子の配置: 吸着種の周囲に明示的に水分子(H2O)を配置して構造最適化を行い、水素結合エネルギーを直接算出します [4, 5]。
- 第一原理分子動力学(AIMD): 水溶媒層を含めた動的なシミュレーションを行い、平均的な安定性を評価します [6](計算コスト:高)。
仮に、図2においてOHの自由エネルギー(青線)を溶媒和効果分(約 -0.3 eV)だけ全体的に下げたと想定すると、0.8 V付近でOH線がO線を下回り、Clean → OH → O という実験と整合する相転移が現れることが定性的に推測できます。
このことは、Pt表面の電気化学反応において、水分子(溶媒)が単なる背景ではなく、表面状態の決定に重要な役割を果たしていることを示唆しています。
まとめ#
本事例では、第一原理計算ソフトウェアAdvance/PHASEを用いて、電気化学環境におけるPt(111)表面の安定性を最もシンプルな真空モデルで評価しました。表面Pourbaix図の計算では、OH相を経由せず直接酸化(O吸着)が進む結果が得られ、これは「水分子によるOHの安定化効果」が考慮されていないことに起因することが分かりました。また、酸化被膜(Pt-O)の形成電位は約0.73 Vと算出され、実験値や詳細な溶媒モデルの結果と概ね一致することから、O原子に対する溶媒効果が限定的であり、本手法が酸化電位の予測に有効であることが示唆されています。
本解析の詳細や、研究への適用可能性に関するご相談はこちら
お問い合わせ参考文献#
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard, and H. Jónsson, "Origin of the overpotential for oxygen reduction at a fuel-cell cathode", J. Phys. Chem. B 108, 17886 (2004).
- J. Rossmeisl, A. Logadottir, and J. K. Nørskov, "Electrolysis of water on (oxidized) metal surfaces", Chem. Phys. 319, 178 (2005).
- D. C. Ford, Y. Xu, and M. Mavrikakis, "Atomic and molecular adsorption on Pt(111)", Surf. Sci. 587, 159 (2005).
- A. Michaelides and P. Hu, "Catalytic Water Formation on Platinum: A First-Principles Study", J. Am. Chem. Soc. 123, 4235 (2001).
- H. A. Hansen, J. Rossmeisl, and J. K. Nørskov, "Surface Pourbaix diagrams and oxygen reduction activity of Pt, Ag and Ni (111) surfaces studied by DFT", Phys. Chem. Chem. Phys. 10, 3722 (2008).
- L. Partanen, and K. Laasonen, "Ab initio molecular dynamics investigation of the Pt (111)–water interface structure in an alkaline environment with high surface OH-coverages", Phys. Chem. Chem. Phy. 26, 18233 (2024).
関連ページ#
- 第一原理計算ソフトウェア Advance/PHASE
- 解析分野:ナノ・バイオ
- 産業分野:材料・化学