遷移金属触媒表面上の硫黄(S)とHSの吸着エネルギーの第一原理解析:スケーリング関係#

硫黄(S)は、触媒化学において極めて重要な元素です。例えば、石油精製における水素化脱硫(HDS)プロセスでは、硫黄化合物の除去が必須であり、その反応メカニズム(S-H結合の切断など)の解明が求められています。一方で、多くの金属触媒(特にPtやPdなど)は、微量の硫黄によって活性を失う「触媒被毒(ポイズニング)」を起こします。この被毒メカニズムや脱硫プロセスの詳細を理解する上で、硫黄(S)とチオラジカル(HS)が触媒表面にどのように吸着し、その吸着エネルギーにどのような関係があるかを知ることは非常に重要です。本解析では、第一原理計算ソフトウェアAdvance/PHASE を用い、いくつかの遷移金属表面におけるSとHSの吸着エネルギーを計算し、両者の間に成り立つスケーリング関係(線形相関性)について解析しました。
Keywords: 第一原理計算 (DFT), 吸着エネルギー, 遷移金属触媒, 硫黄(S), HS, 触媒被毒, 水素化脱硫(HDS), スケーリング関係
計算モデルと計算条件#
本解析では、DFT計算対象の遷移金属として Ir, Pt, Pd, Cu, Ag, Au を選定しました。まず、各金属のバルク構造について図1で示したように格子定数を最適化し、その格子定数を用いて表面スラブモデルを構築しました。スラブモデルは、2x2の表面スーパーセルを用い、3層の原子層(底面2層は固定)で構成し、表面間の相互作用を避けるために 15 Å の真空層を設けました。
S原子の吸着については、表面の主要な高対称性サイト(top, bridge, fcc hollow, hcp hollow)に原子を配置し、構造最適化を行って最安定な吸着サイトを決定しました。HS分子の吸着については、吸着サイトだけでなく、S-H結合の向き(表面に垂直、斜めなど)もエネルギーに影響するため、様々な初期配置からの構造最適化を行いました。
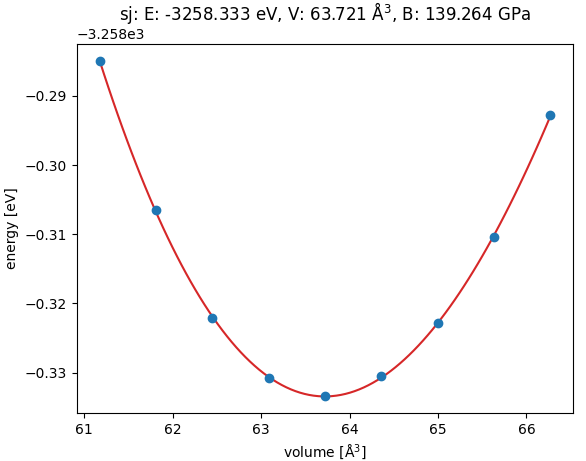
図1. fcc PdのE-V曲線(エネルギー対体積)。Vは慣用セルの体積です。この計算から、格子定数は3.9942 Åと決定されました。交換相関汎関数RPBEは吸着計算に適していますが、格子定数を過大評価する傾向があるため、スラブ計算にはRPBEで最適化した一貫性のある格子定数を使用しました。
本解析で用いた主な計算条件は表1に示されています。
表1. 主な計算条件
| 項目 | 設定 |
|---|---|
| 擬ポテンシャル | ウルトラソフト (H: ノルム保存) |
| 交換相関汎関数 | RPBE |
| 波動関数のカットオフエネルギー | 25 ~ 50 Rydberg |
| 表面スーパーセル | 2x2 |
| 原子層 | 3層(底面2層固定) |
| 真空層 | 15 Å |
計算結果と考察#
吸着エネルギーの定義#
吸着エネルギーは、吸着後の系全体のエネルギーから、吸着前のクリーンな表面(スラブ)と吸着種のエネルギーの和を差し引くことで定義されます。S原子の吸着エネルギー は以下の式で計算されます。
- : S原子が表面に吸着した系全体のエネルギー
- : クリーンなスラブ単体のエネルギー
- : 参照状態として、真空中に置かれたS原子1個のエネルギー
同様に、HS分子の吸着エネルギー は以下の式で定義されます。
- : HS分子が表面に吸着した系全体のエネルギー
- : クリーンなスラブ単体のエネルギー(上記と同じ)
- : 参照状態として、真空中に置かれたHS分子(ラジカル)1個のエネルギー
最安定な吸着構造#
構造最適化の結果、得られた最安定な吸着構造の例(Pd表面)を図2に示します。

図2. Pd表面におけるHS(左)とS(右)の最安定吸着構造(上面図)。
S原子の吸着では、計算した全ての金属表面において、3つの金属原子に囲まれた fcc hollow サイトが最安定となりました。一方、HS分子の吸着では、S原子が2つの金属原子に結合する bridge サイトに近い位置で、S-H結合が表面に対して平行に近い、わずかに傾いた配置が安定となりました。このbridgeサイトに吸着する構造は、文献 [1] によるPd(111)表面上のHS吸着に関するDFT研究の結果とも一致しています。
吸着エネルギーのスケーリング関係#
計算されたSの吸着エネルギー を横軸に、HSの吸着エネルギー を縦軸にとり、各金属(Ir, Pt, Pd, Cu, Ag, Au)のデータをプロットしたものが図3です。
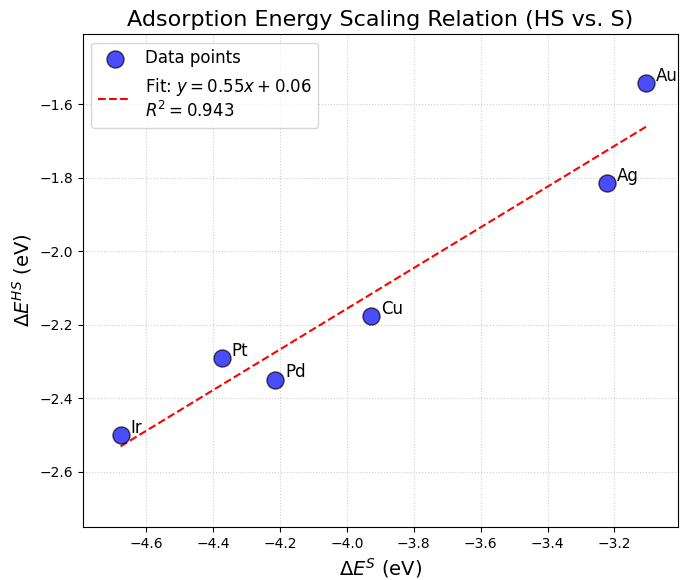
図3. Sの吸着エネルギー () とHSの吸着エネルギー () のスケーリング関係。データ点(青丸)は各金属表面での計算値を示し、赤色の破線は線形フィッティングの結果です。
図3から明らかなように、 と の間には強い線形相関があることがわかります。フィッティングによって得られた関係式は であり、決定係数 も 0.943 と高い値を示しました。これは、S原子が強く吸着する金属( が負に大きい、例:Ir)ほど、HS分子もまた強く吸着する傾向があることを定量的に示しています。
理論的予測との比較#
得られた線形関係 は、Nørskovらによって提唱された触媒表面における吸着エネルギーの一般化されたスケーリング則 [2] と比較することができます。
この理論によれば、 という化学種の吸着エネルギー は、中心原子 A の吸着エネルギー と以下の線形関係で近似されます。
ここで、傾き は、中心原子Aが結合できる最大のH原子数 と、現在のH原子数 を用いて、 で与えられます。硫黄(S)の場合、安定な 分子を考慮すると とみなすことができます。我々が計算している 分子は に相当します。したがって、理論的に予測される傾き は、
となります。本研究で得られた傾き 0.55 は、この理論的予測値 0.5 と良好な一致を示しており、計算結果の妥当性と、S/HS吸着系がスケーリング則によく従うことを裏付けています。
スケーリング則の有用性#
本解析で示されたようなスケーリング関係は、触媒研究において非常に強力なツールとなります。その最大の有用性は、計算コストの削減と触媒設計の加速にあります [3, 4]。通常、触媒反応のメカニズムを解明するには、反応に関わるすべての中間体(HS, S, Hなど)の吸着エネルギーを個別に計算する必要があり、多大な計算リソースを要します。しかし、 と のように中間体同士の吸着エネルギーに線形な相関(スケーリング則)が成り立てば、より計算が単純な (あるいは や といった原子の吸着エネルギー)を計算するだけで、複雑な中間体( など)の吸着エネルギーを高い精度で予測することが可能になります。これにより、無数の候補材料に対して網羅的な計算を行うことなく、有望な触媒を迅速にスクリーニング(探索)することが可能となり、新しい触媒材料の設計・開発が大幅に加速されると期待されます。
補足:スケーリング則から「外れる」要因について
本文では と の間に強い線形相関が見られましたが、プロットを詳細に見ると、PdやAuなどはフィットラインからわずかに外れています。この「スケーリング則からの外れ」は、単なる誤差ではなく、重要な物理的・化学的意味を持つと考えられます。
主な要因としては、以下の2点が挙げられます。
- 吸着サイトの違い(幾何学的要因): 本解析でも示されたように、S原子はhollowサイト、HS分子はbridgeサイトを最安定とします。金属種によって、これら異なるサイトに対する親和性のバランスが微妙に異なるため、単純な線形関係からのズレが生じる可能性があります。
- 電子状態の特異性(化学的要因): スケーリング則は、SとHSの吸着が主に金属のdバンドとの相互作用という共通の要因で決まることを前提としています。しかし、HS分子はS原子にはないS-H結合の軌道(軌道や軌道)を持っており、特定の金属表面がこの軌道と特異的に強く相互作用する場合(例:強いバックドネーション)、HSは予測よりも余計に安定化され、ラインから外れることになります。
このように「外れ」の要因を分析することは、各金属が持つ触媒としての「個性」や「選択性」(特定の結合を活性化しやすい、など)を理解する上で重要な手がかりとなります。
まとめ#
本解析では、第一原理計算ソフトウェアAdvance/PHASE を用い、遷移金属(Ir, Pt, Pd, Cu, Ag, Au)表面上における硫黄(S)およびチオラジカル(HS)の吸着エネルギーを計算しました。S原子はfcc hollowサイト、HS分子はS原子がbridgeサイトに近い位置において傾いた配置で最も安定に吸着することがわかりました。さらに、Sの吸着エネルギー とHSの吸着エネルギー の間には、強い線形相関(スケーリング関係)が存在することを見出しました。その傾きは、Nørskovらによる一般化されたスケーリング則の理論予測値とよい一致を示しました。このようなスケーリング関係の定式化は、触媒被毒や水素化脱硫といった複雑な触媒プロセスを理解し、計算化学に基づいた合理的な触媒設計を加速するための重要な知見となります。
本解析の詳細や、研究への適用可能性に関するご相談はこちら
お問い合わせ参考文献#
- M. P. Hyman, B. T. Loveless, and J. W. Medlin, "A density functional theory study of H2S decomposition on the (111) surfaces of model Pd-alloys", Surf. Sci. 601, 5382 (2007).
- F. Abild-Pedersen, J. Greeley, F. Studt, J. Rossmeisl, T. R. Munter, P. G. Moses, E. Skúlason, T. Bligaard, and J. K. Nørskov, "Scaling Properties of Adsorption Energies for Hydrogen-Containing Molecules on Transition-Metal Surfaces", Phys. Rev. Lett. 99, 016105 (2007).
- J. K. Nørskov, T. Bligaard, J. Rossmeisl, and C. H. Christensen, "Towards the computational design of solid catalysts", Nat. Chem. 1, 37 (2009).
- J. Greeley, "Theoretical Heterogeneous Catalysis: Scaling Relationships and Computational Catalyst Design", Annu. Rev. Chem. Biomol. Eng. 7, 605 (2016).
関連ページ#
- 第一原理計算ソフトウェア Advance/PHASE
- 解析分野:ナノ・バイオ
- 産業分野:材料・化学