DFT計算をエンジンとした自動構造探索:Optunaによる表面吸着サイトの効率的同定#

触媒材料やガスセンサーの設計において、固体表面への分子吸着挙動を原子スケールで理解することは不可欠です。しかし、吸着位置()や高さ()、さらに分子の配向(角度)といった多次元のパラメータ空間から最安定構造を特定するには、膨大な数の計算が必要となり、計算コストが大きな障壁となります。本解析では、第一原理計算ソフトウェアAdvance/PHASEをエネルギー評価エンジン(DFT計算)とし、ハイパーパラメータ自動最適化フレームワーク「Optuna」と連携させることで、白金(Pt)表面へのアンモニア(NH3)吸着サイトをハイスループットかつ高精度に同定する計算パイプラインを構築しました。
Keywords: 第一原理計算 (DFT), Optuna, ベイズ最適化, 表面吸着, アンモニア (NH3), Pt(111), 解析自動化, マテリアルズ・インフォマティクス
解析手法と計算ワークフロー#
最適化フレームワーク「Optuna」の導入#
本解析の核となる構造探索には、機械学習向けに設計された、オープンソースの最適化フレームワークであるOptuna [1] を活用しました。Optunaは、未知の目的関数の形状を過去の試行結果から確率的に推定する「ベイズ最適化」を実装しています。特に本解析で採用したTPE (Tree-structured Parzen Estimator)アルゴリズムは、高次元の探索空間において「有望な領域(Exploitation:利用)」の深掘りと「未知の領域(Exploration:探索)」の調査をバランスよく行います。
表面吸着系において、原子配置や回転角といった連続変数と、配向の上下といったカテゴリカル変数が混在する複雑なポテンシャルエネルギー曲面に対して、Optunaはランダムサーチやグリッドサーチを凌駕する効率で、グローバルミニマム(最安定点)へと収束する能力が期待されます。これにより、主観的な初期構造選定に依存せず、バイアスのない構造探索が可能となります。
2段階の自動探索ワークフロー#
以下のワークフローはASE(Atomic Simulation Environment)[2] を利用して構築しました。なお、PHASE専用GUIであるPHASE-Viewerでも構造最適化の入力設定・ジョブ実行・結果解析が可能です。
- 第1段階:Global Coarse Search(Optunaによる広域探索) スラブモデルの原子を固定した状態で、Optunaを用いて、NH3の吸着位置(分数座標 )、表面からの高さ()、および分子の向き(上下配向・傾き・回転)をハイパーパラメータとして探索しました。各試行ではAdvance/PHASEによる単点エネルギー計算(SCF)を実行し、200回程度の試行を通じてポテンシャルエネルギー曲面の概形を把握しました。
- 第2段階:Local Fine Optimization(局所構造最適化) 第1段階で見つかった最安定候補構造に対し、表面上部2層および分子を可動とした構造最適化(BFGS法)を実施しました。これにより、表面緩和を含む最終的な安定構造を確定させました。
計算条件:
- モデル:Pt(111) 2x2 スラブ(3層、真空層15 Å)
- 格子定数(バルク): Å(RPBE + DFT-D3にて事前最適化済み)
- 汎関数:RPBE + DFT-D3(ファンデルワールス補正)
- 擬ポテンシャル:ウルトラソフト型擬ポテンシャル(H: ノルム保存型)
計算結果と考察#
1. Optunaによる探索プロセスの可視化#
図1に、Optunaによる全試行の探索履歴に基づくポテンシャルエネルギー曲面(PES)の投影図を示します。
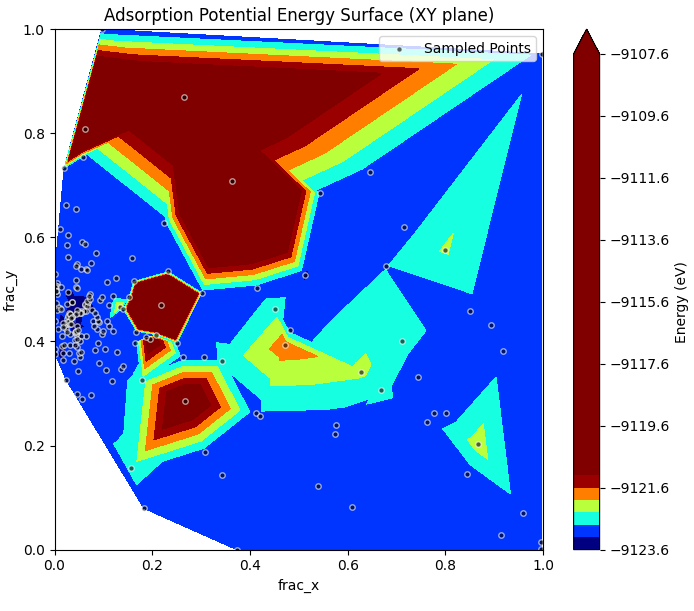
図1. Pt(111) 2×2スラブモデル内における吸着位置(分数座標 \(x, y\))と全エネルギーの関係。白枠の黒点はOptunaがサンプリングした計算ポイント。ベイズ最適化により、安定領域(Topサイト付近)へ集中的に計算リソースが投入されていることが分かります。
図1において最大で約16 eVもの巨大なエネルギー変動が見られますが、これは探索空間に『表面からの高さ()』や『分子の配向』が含まれていることに起因します。第1段階は構造緩和を伴わないSCF単点計算であるため、Optunaが生成した乱数によっては、吸着分子が表面原子に近すぎる(めり込む)ような非物理的な初期構造が生成されることがあります。このような強い立体反発(パウリ反発)による高いエネルギー状態もアルゴリズムが『不正解』として正しく学習し、効率的に安定構造へ探索を絞り込めていることが分かります。この図から、Optunaが序盤の探索を通じて低エネルギー領域(青色の谷底)を学習し、最安定サイトであるTopサイト(Pt原子の直上)周辺を重点的に探索している様子が確認できます。従来のグリッドサーチ(総当たり)に比べて、ベイズ最適化を用いることでより効率的に正解へ到達しました。
※図1の等高線マップは、Optunaによる非均一なサンプリング点(白枠の黒点)を元に補間描画したものです。網羅的なグリッドサーチとは異なり、序盤で高エネルギー領域(赤色)の探索を打ち切り、低エネルギーの最安定サイト周辺(青色)に計算リソースを集中させています。これは『ベイズ最適化特有の挙動』を示しており、結晶表面本来の周期的な対称性ではなく、探索の軌跡が強く反映された図となっています。
2. 最安定構造と吸着配向#
Optunaを用いた探索の結果、Pt(111)のTopサイトに「窒素(N)が表面側を向く配向(N-down)」をエネルギーの一番低い構造として特定しました。図2に、第2段階の構造最適化を経て得られた最終的な吸着構造を示します。この吸着構造は先行研究 [3] の計算結果とも整合しています。
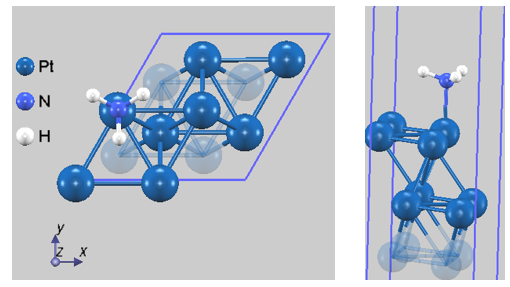
図2. 最適化されたNH3/Pt(111)吸着構造。窒素の非共有電子対がPt原子へ配位し、水素原子が上方に広がる傘型の安定構造が精度良く再現されています(Pt-N結合長: 2.158 Å)。
3. 吸着エネルギーの評価#
最終的な構造最適化結果から、吸着プロセスの安定性を定量化する指標として吸着エネルギー を算出しました。 は、孤立状態の分子が表面に吸着した際の系のエネルギー変化を表し、以下の式で定義されます。
- :分子が吸着した全体の系のエネルギー(第2段階の結果)
- :Pt(111)表面スラブ単体のエネルギー
- :真空中に孤立したNH3分子単体のエネルギー
本解析において、すべての項を同一の汎関数および計算条件にて算出した結果、吸着エネルギーは -0.83 eV となりました。この負の値は吸着が発熱的であることを示しています。定量的な妥当性を評価すると、本解析の値(RPBE + DFT-D3:-0.83 eV)は、先行研究 [3] におけるGGA-PW91計算値(-0.78 eV)と整合するだけでなく、実験的に求められた吸着エネルギー(-0.82 eV)[4] と非常に良好な一致を示しています。これは、本自動探索ワークフローが単なる効率化に留まらず、高精度な物理量を導出できていることを裏付けています。
まとめ#
本事例では、第一原理計算ソフトウェアAdvance/PHASEとOptunaを連携させ、複雑な表面吸着構造探索を大幅に効率化しました。本手法の最大の利点は、「物理化学的な事前知識なしに、機械学習ベースの探索アルゴリズムが安定な配向やサイトを自動的に学習・特定できる点」にあります。これにより、主観や手作業による試行錯誤を排除し、ハイスループットかつ信頼性の高い解析が可能となります。このワークフローは、触媒の反応機構解析、二次電池の電極界面設計、半導体表面の分子修飾など、広範なマテリアルズ・インフォマティクス分野への応用が期待されます。
本解析の詳細や、研究への適用可能性に関するご相談はこちら
お問い合わせ参考文献#
- T. Akiba, S. Sano, T. Yanase, T. Ohta, and M. Koyama, "Optuna: A Next-generation Hyperparameter Optimization Framework", Proceedings of the 25th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, 2623 (2019).
- A. H. Larsen et al., "The Atomic Simulation Environment—A Python library for working with atoms", J. Phys.: Condens. Matter 29, 273002 (2017).
- D. C. Ford, Y. Xu, and M. Mavrikakis, "Atomic and molecular adsorption on Pt(111)", Surf. Sci. 587, 159 (2005).
- J. L. Gland, "Adsorption and decomposition of nitric oxide and ammonia on a stepped platinum single crystal surface", Surf. Sci. 71, 327 (1978).
関連ページ#
- 第一原理計算ソフトウェア Advance/PHASE
- 解析分野:ナノ・バイオ
- 産業分野:材料・化学