触媒による不飽和結合の活性化:Pt(111)表面上のC2H4吸着#

エチレン(エテン、C2H4)の水素化反応は、工業的に極めて重要な不飽和炭化水素の転換反応のモデルとして、長年にわたり研究されてきました。特に白金(Pt)は優れた触媒活性を示しますが、その反応メカニズムを原子レベルで解明することは、より高効率な触媒設計に不可欠です。本解析では、第一原理計算ソフトウェアAdvance/PHASEを用い、Ptの代表的な表面である(111)面上でのエチレンの吸着挙動をシミュレーションしました。吸着エネルギーや安定構造、さらには電子のやり取りを可視化することで、触媒作用の根源となる化学結合の本質に迫ります。
Keywords: 第一原理計算, DFTシミュレーション, C2H4, Pt(111), 吸着エネルギー, 差分電荷密度, di-σ結合, π結合, van der Waals補正
計算モデルと計算条件#
本解析では、まずPtバルク結晶の構造を最適化し、格子定数として約4.00 Åを得ました。この最適化されたバルク構造から、Pt(111)表面を模擬した2x2の表面スーパーセルモデル(4層および6層のスラブモデル)を作成し、その上に1分子のエチレンを配置しました(図1参照)。計算コストと精度を両立させるため、スラブの下半分(2層または3層)はバルクの位置に固定しました。
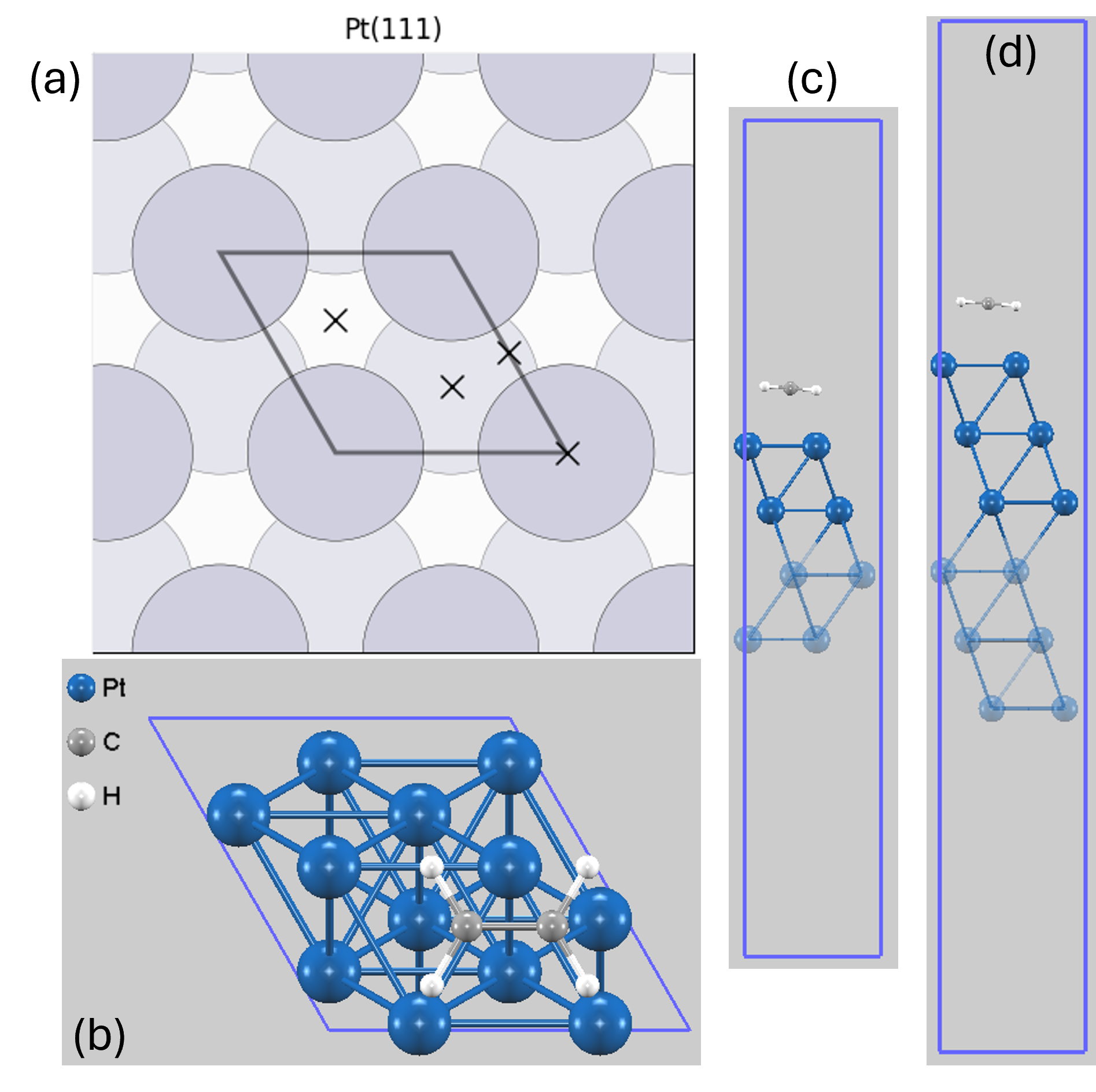
図1. (a) Pt(111)表面の代表的な吸着サイト(×印): トップサイト (on-top)、 ブリッジサイト (bridge)、 ホローサイト (fcc/hcp-hollow)、(b) C2H4の初期配置例(上面図)、(c) 4層スラブモデル、(d) 6層スラブモデル。透明な原子は計算中に固定されています。
表面上の分子吸着は、吸着サイトだけでなく、分子の向きや配置によっても安定性が大きく変わります。第一原理計算における構造最適化は、通常、与えられた初期構造に最も近い局所安定構造を探索します。そのため、物理的に妥当ないくつかの初期配置から計算を開始し、最もエネルギー的に安定な構造(最安定構造)を見つけ出すことが重要です。本解析では、過去の知見 [1-6] に基づき、エチレン分子が表面にほぼ平行に配向した構造を初期配置として採用しました。
表1. 計算条件の概要
| 項目 | 設定 |
|---|---|
| 擬ポテンシャル | ウルトラソフト擬ポテンシャル (H: ノルム保存) |
| 交換相関汎関数 | GGA (PBE) |
| van der Waals補正 | DFT-D3 |
| カットオフエネルギー | 25 Rydberg (約340 eV) |
| k点サンプリング | 7x7x1 |
計算結果と考察#
安定な吸着構造#
様々な初期配置から構造最適化を行った結果、Pt(111)表面上には主に2種類の安定な吸着構造が存在することが明らかになりました(図2)。
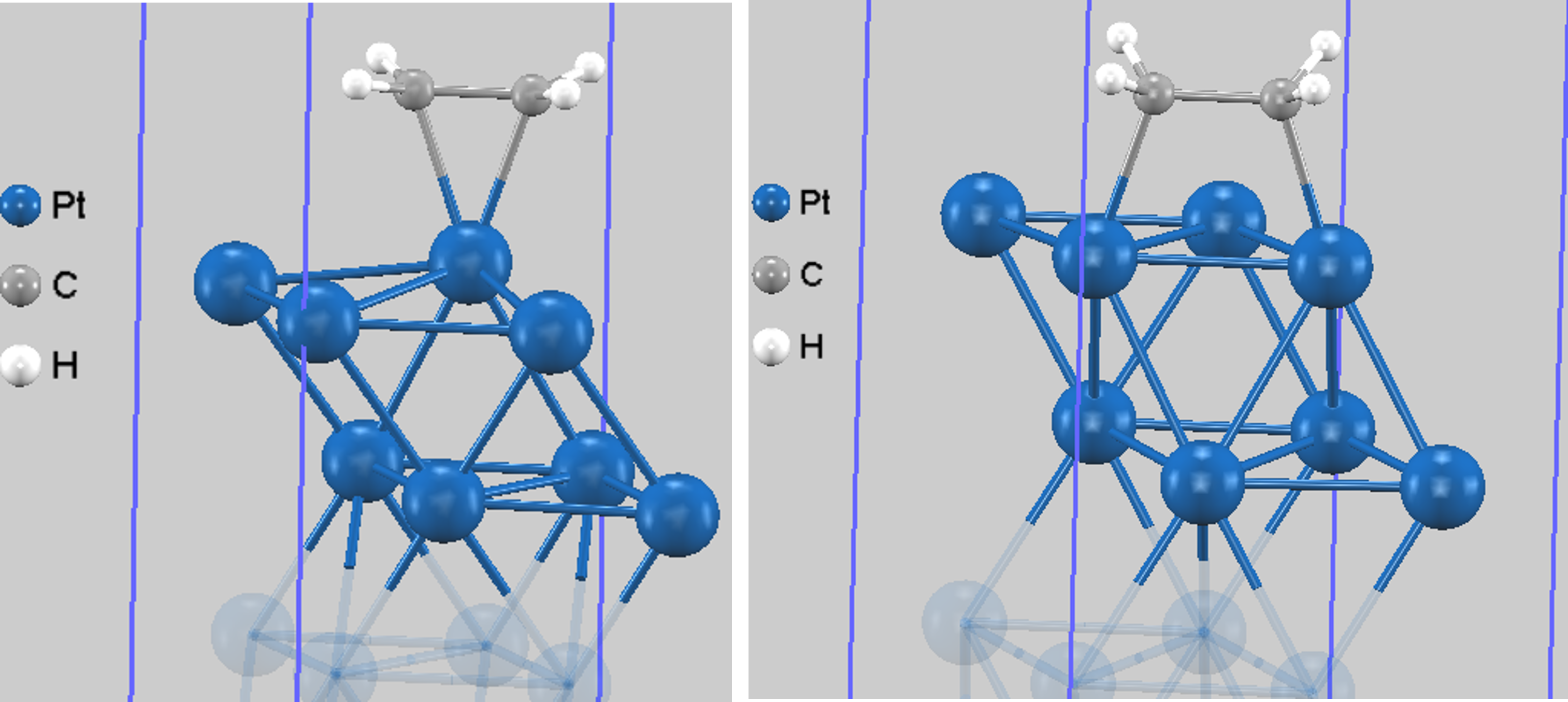
図2. 構造最適化で得られた安定構造。左: 安定構造Ⅰ (π結合型)。Pt原子の真上(トップサイト)に吸着。C-C結合長は約1.42 Å。 右:安定構造Ⅱ (di-σ結合型)。2つのPt原子の間(ブリッジサイト)に吸着。C-C結合長は約1.50 Åと大きく伸長し、水素原子が面外に折れ曲がります。
構造Ⅰは、エチレンのπ電子雲とPt原子が相互作用する「π結合型」吸着に対応します。一方、構造Ⅱは、エチレンのC=C二重結合が切れ、2つのC原子がそれぞれPt原子と新しいσ結合を形成する「di-σ結合型」吸着に対応します。di-σ型は、C-C結合がより単結合に近くなり、分子の変形が大きいことが特徴です。これは、吸着に伴い炭素原子の混成軌道が、平面構造のsp2混成から、より立体的なsp3混成へと変化する「再混成」が起きるためです。
吸着エネルギーと構造の比較#
吸着エネルギーは、分子が表面に吸着する際の安定化の度合いを示す指標です。以下の式で定義されます。
ここで、は吸着後の全系のエネルギー、は表面単体のエネルギー、は分子単体のエネルギーです。
計算されたC-C結合長と吸着エネルギーを、スラブの層数やvan der Waals (vdW)補正の有無で比較し、さらに既存の実験値と対照しました(表2、表3)。
表2. 吸着構造(C-C結合長)の比較 (単位: Å)
| 構造 | 4層, GGA | 6層, GGA | 6層, GGA+D3 | 実験値 [4, 5] |
|---|---|---|---|---|
| π結合型 | 1.42 | 1.42 | 1.42 | 1.41 |
| di-σ結合型 | 1.50 | 1.50 | 1.50 | 1.48-1.52 |
表3. 吸着エネルギーの比較 (単位: kJ/mol)
| 構造 | 4層, GGA | 6層, GGA | 6層, GGA+D3 | 実験値 [5] |
|---|---|---|---|---|
| π結合型 | -69 | -67 | -115 | -40 ± 10 |
| di-σ結合型 | -104 | -108 | -154 | -38 ~ -71 |
考察:
- モデルの妥当性: 4層モデルと6層モデルの結果に大きな差異はなく、4層スラブモデルは計算コストと精度を両立できる良いモデルであることが確認できました。
- 構造の再現性: 計算で得られたC-C結合長は、いずれの条件でも実験値の範囲と非常によく一致しており、2つの吸着状態の幾何構造を正確に再現できていることがわかります。
- エネルギーの傾向と他のDFT計算との比較: 全ての計算条件で、di-σ結合型の方がπ結合型よりもエネルギー的に安定であるという結果が得られました。これは、より多くの、そしてより強い結合が形成されるためです。過去のGGA汎関数を用いたDFT計算では、di-σ型の吸着エネルギーは-100 kJ/mol [1] から-127 kJ/mol [4] の範囲で報告されており、本解析のGGA計算値(-104, -108 kJ/mol)もこの傾向と一致しています。
- vdW補正の影響: vdW補正(DFT-D3)を考慮すると、吸着エネルギーが46 kJ/molほど大幅に増加(より安定化)しました。しかし、GGA+D3による計算値は実験値よりもかなり大きく、この系では過大評価の傾向があることが示唆されます。他の研究でも、RPBE汎関数にTkatchenko–Scheffler法のvdW補正を加えると、この系の吸着エンタルピーが43 kJ/molほど増加して、過大評価になる可能性が指摘されております [2]。これまでの研究と同様に、本解析でvdW補正なしのGGA(PBE)計算値の方が、実験値の範囲により近い結果となりました。
補足:
- vdW補正と誤差の相殺について: 一般的なGGA汎関数(本解析ではPBE)は、vdW相互作用を記述できない一方で、交換反発を不正確に記述するという性質を持ちます。vdw相互作用が重要となる系で、vdW補正なしの計算値が実験値と見かけ上よく一致する現象は、これら二つの異なる誤差が偶然打ち消し合った結果(誤差の相殺)である可能性が指摘されています。物理的な妥当性の観点からは、vdW補正を含めることが金属表面における分子吸着計算では一般的により正しいアプローチとなります。
- 計算値と実験値の比較について: 本計算の吸着エネルギーは絶対零度(0K)・真空中での理論値です。一方、実験で得られる吸着エンタルピーは、有限な温度・圧力下で測定され、零点振動エネルギーやエントロピーの効果を含みます。そのため、両者を直接定量的に比較する際には、これらの前提条件の違いを考慮する必要があります。
差分電荷密度解析:化学結合の可視化#
分子が表面に吸着する際に電子がどのように移動したかを可視化するため、差分電荷密度解析を行いました。これは、3つの独立した計算結果を組み合わせることで、吸着による電子の再配置のみを抽出する手法です。
計算手順:
- 全体系の計算:まず、エチレンが吸着したPt(111)スラブモデル全体の構造最適化を行い、最安定構造とその電荷密度 を求めます。
- スラブ単体の計算:次に、ステップ1で得られた最安定構造と全く同じ原子座標にPt原子を固定した状態で、Ptスラブ単体の電荷密度 を計算します。
- 分子単体の計算:同様に、ステップ1の最安定構造と全く同じ原子座標にC原子とH原子を固定した状態で、エチレン分子単体の電荷密度 を計算します。
- 差分の算出:最後に、ステップ1の電荷密度からステップ2と3の電荷密度の和を差し引くことで、吸着相互作用に起因する電荷の移動分 を得ます。
図3では、di-σ型吸着構造において、電子が増加した領域(電子蓄積)が赤、減少した領域(電子枯渇)が青で示されています。4層モデル (GGA)で得られた結果は、6層モデル (GGA+D3)で得られた結果と非常に良く一致しています。
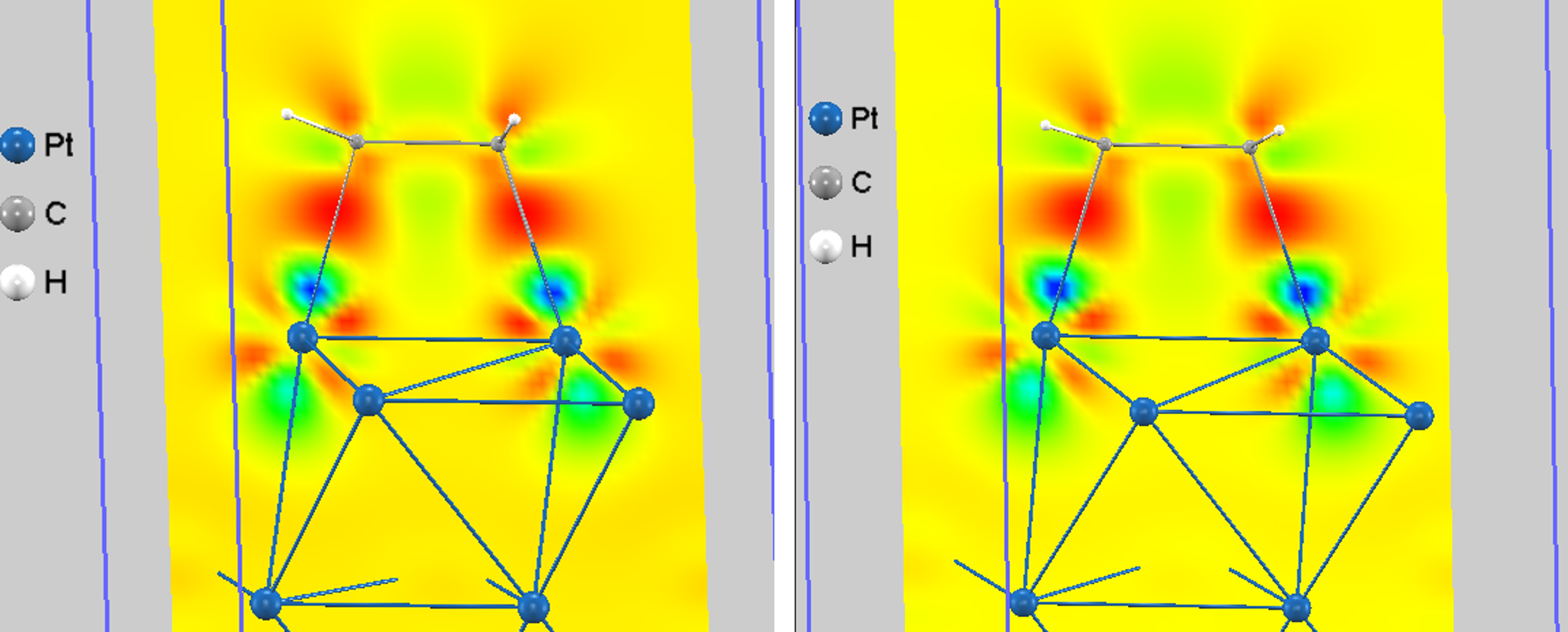
図3. di-σ型吸着構造における差分電荷密度(左:4層モデルでGGA使用、右:6層モデルでGGA+D3使用)。赤は電子の増加、青は電子の減少を示します。
図3から、以下の化学結合メカニズムが明らかになります。
- Pt-C間の強い結合形成: Pt原子とC原子の間に顕著な赤い領域(電子蓄積)が見られ、2つの強固なPt-C共有結合が形成されたことがわかります。
- C=C π結合の消失: C-C結合軸の上下にあったπ電子(青い領域、電子枯渇)が、Pt-C結合の形成のためにPt原子へ供与されました。
- Ptからの電子の逆供与: Pt原子周辺の一部領域(青い領域)は電子密度が減少していることが見られ、これはPt原子が結合形成に電子を拠出していることを示唆します。この現象は、Ptのd軌道電子がエチレンのπ*反結合性軌道へ返される「逆供与」として知られており、Pt-C間の結合をさらに強化する働きをします [5]。
- C-C σ結合の維持: 2つの炭素原子間には赤い領域が残っており、分子骨格であるC-C単結合(σ結合)は維持されています。
これらの電子のやり取りにより、エチレンの炭素原子とPt表面の原子の間に、それぞれ独立した2つの新しいσ結合が形成されます。このため、この吸着形態は「di-σ結合」と呼ばれます。
まとめ#
本解析では、第一原理計算ソフトウェアAdvance/PHASEを用いてPt(111)表面上でのエチレンの吸着をシミュレーションし、di-σ結合型とπ結合型の2つの安定な吸着形態を特定しました。計算で得られた吸着構造は実験値と非常によく一致し、di-σ型がよりエネルギー的に安定であることが示されました。van der Waals補正(DFT-D3)は吸着エネルギーを実験値より過大評価する傾向がありましたが、差分電荷密度解析により、di-σ結合がエチレンのπ電子の供与とPtからの電子の逆供与によって形成される2つの強固なPt-C結合であることが視覚的に明らかになりました。これらの結果は、第一原理計算が触媒表面での化学吸着メカニズムを原子・電子レベルで解明する上で強力なツールであることを示しています。
本解析の詳細や、研究への適用可能性に関するご相談はこちら
お問い合わせ参考文献#
- J. M. Essen, J. Haubrich, C. Becker, and K. Wandelt, "Adsorption of ethene on Pt(111) and ordered PtxSn/Pt(111) surface alloys: A comparative HREELS and DFT investigation", Surf. Sci. 601, 3472 (2007).
- R. Miyazaki, S. Faraji, S. V. Levchenko, L. Foppa, and M. Scheffler, "Vibrational frequencies utilized for the assessment of exchange–correlation functionals in the description of metal-adsorbate systems: C2H2 and C2H4 on transition-metal surfaces", Catal. Sci. Technol. 14, 6924 (2024).
- R. M. Watwe, R. D. Cortright, M. Mavrikakis, J. K. Nørskov, and J. A. Dumesic, "Density functional theory studies of the adsorption of ethylene and oxygen on Pt(111) and Pt₃Sn(111)", J. Chem. Phys. 114, 4663 (2001).
- Z.-J. Zhao, L. V. Moskaleva, H. A. Aleksandrov, D. Basaran, N. Rösch, "Ethylidyne formation from ethylene over Pt(111): A mechanistic study from first-principle calculations", J. Phys. Chem. C 114, 12190 (2010).
- Q. Ge, and D. A. King, "The chemisorption and dissociation of ethylene on Pt{111} from first principles", J. Chem. Phys. 110, 4699 (1999).
- T. Okada, Y. Kim, Y. Sainoo, T. Komeda, M. Trenary, and M. Kawai, "Coexistence and interconversion of di-σ and π-bonded ethylene on the Pt(111) and Pd(110) surfaces", J. Phys. Chem. Lett. 2, 2263 (2011).
関連ページ#
- 第一原理計算ソフトウェア Advance/PHASE
- 解析分野:ナノ・バイオ
- 産業分野:材料・化学