貴金属エッチングの第一原理シミュレーション:配位子設計と面異方性の熱力学解析#

金(Au)などの貴金属は極めて化学的安定性が高いため、従来のエッチング工程ではシアン化物や王水といった毒性の強い薬剤が主に使用されてきました。環境負荷の低減と微細加工精度の向上を両立させるため、ヨウ素系やチオ尿素系といった代替エッチャントの開発が求められています。本事例では、第一原理計算ソフトウェアAdvance/PHASEによる表面エネルギー評価と、NWChemによる溶液錯体化学解析を連携させ、配位子の種類と結晶面方位がエッチング性能に与える影響を定量的に評価しました。
Keywords: 第一原理計算 (DFT), Auエッチング, 配位子設計, 表面エネルギー, 熱力学安定性, 異方性エッチング
解析手法と理論背景#
エッチング反応は、「表面の金属原子が酸化される過程」と「溶液中の配位子によって錯体として安定化し溶解する過程」の組み合わせで構成されます。本解析では、バルク状態のAuから1原子が離脱し、溶液中で錯体を形成するまでの自由エネルギー変化 を算出しました。
エッチングエネルギーの評価指標
- 反応式: 共通の酸化剤としてヨウ素分子()を想定し、以下の反応を評価します [1]。 (※チオ尿素のような中性配位子の場合は、生成する錯体は となります)
- エッチングエネルギーの算出: 反応式に従って、Auの凝集エネルギー()と、Advance/PHASEで算出した表面エネルギー()を、NWChem [2] による錯体・反応物・孤立原子のエネルギー()に統合しました。これにより、基礎エッチングエネルギー()と表面ごとの実効エッチングエネルギー()を算出しました。
- 計算条件: NWChemを用いたDFT計算において、重原子(Au, I)には有効コアポテンシャル(ECP: LANL2DZ)を適用し、水溶液環境を再現するために連続誘電体モデル(COSMO)を使用しました。
Advance/PHASEによるAu表面の熱力学的評価#
エッチングの「削れやすさ」の起点となる結晶面の不安定性を評価するため、Advance/PHASEを用いて各方位の表面エネルギーを算出しました。格子定数はDFT計算値である 4.173 Å を使用し、単位面積当たりのエネルギーから表面原子1個当たりの不安定化エネルギーへと換算しました。
表1. Advance/PHASEで算出したAu各結晶面の特性。
| 結晶面 | 表面エネルギー [J/m²] | 不安定化エネルギー [eV/atom] | 特徴 |
|---|---|---|---|
| Au(111) | 0.69 | 0.325 | 最密充填面。熱力学的に最も安定で削れにくい。 |
| Au(100) | 0.87 | 0.473 | (111)面に比べ原子配列が疎で、反応の起点になりやすい。 |
| Au(110) recon. | 0.84 | 0.646 | 再構成構造。表面原子1個当たりの不安定性が(111)の約2倍。 |
NWChemによる配位子・錯体の安定性解析#
代表的な配位子であるシアン(CN⁻)、チオ尿素、ヨウ素(I⁻)、塩素(Cl⁻)について、NWChem(B3LYP/6-311+G**、もしくはB3LYP/LANL2DZ)を用いて、水溶液中における金錯体 の形成エネルギーを算出しました。ここで、計算誤差を補正し実験事実との整合性を図るため、凝集エネルギーには信頼性の高い実験値 eV を採用しました。これにより、固体状態からの基準反応エネルギー を導出し、以下の表面エネルギー解析と組み合わせました。
結果と考察#
1. 配位子ごとの基礎エッチング能力と実験事実の再現#
まず、バルク状態のAuに対する基礎エッチングエネルギー()を算出し、配位子間の優劣を比較しました(表2)。
表2. 各配位子における基礎エッチングエネルギー(\(\Delta E_\text{bulk}\))。
| 配位子系 | [eV] | 判定 |
|---|---|---|
| シアン (CN⁻) | -1.271 | 自発的(非常に強力) |
| チオ尿素 | +0.393 | 条件により進行(実用的) |
| ヨウ素 (I⁻) | +0.536 | 条件により進行(マイルド) |
| 塩素 (Cl⁻) | +0.998 | 非自発的(単独溶解不可) |
計算結果から、塩素系は基礎エッチングエネルギーが大きな正(吸熱)となり、「塩化物イオン単独では金を溶解できない」という実験事実が裏付けられました。対照的に、シアン系はが -1.271 eV と大きな負の値となっており、自発的に溶解が進行する強力なエッチャントであることが示されました。
2. チオ尿素・ヨウ素が示す「面選択的(異方性)エッチング」のメカニズム#
次に、Advance/PHASEで得た各面の不安定化エネルギーを適用し、実効エッチングエネルギーの面方位依存性を評価しました(図1)。
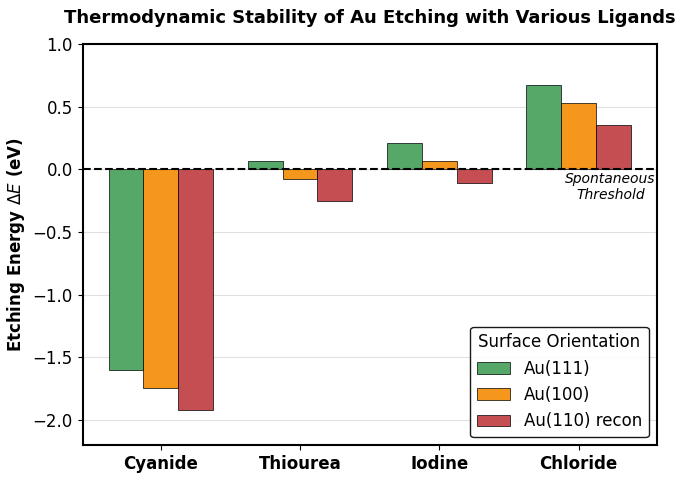
図1. 結晶面ごとの実効エッチングエネルギーの変化。チオ尿素とヨウ素は、不安定な面でのみ自発的溶解(ゼロ以下)を示します。※グラフのゼロ点は酸化剤の酸化還元電位に依存しています。
安定な(111)面に対してはエネルギー障壁が正(溶けにくい)であるのに対し、より不安定な(110)面では両配位子ともにエネルギーが負(自発的溶解)に転じます。また(100)面においても、チオ尿素で負に転じるほか、ヨウ素でも自発的溶解の閾値付近まで障壁が大幅に低下することが確認できます。この結果は、これらのマイルドな配位子が「平坦なテラス面(111)を維持しつつ、よりエネルギー的に不安定な平面(110, 100等)、特にステップや欠陥などの活性サイトを持つ高エネルギー面から優先的にエッチングを進行させる」という、異方性エッチングや表面平滑化の物理的メカニズムを明確に示唆しています。
3. ミクロな反応機構:チオ尿素の吸着アンカー特定#
マイルドなエッチャントとして実用性が示されたチオ尿素について、NWChemを用いて水溶液中(COSMOモデル)における静電ポテンシャル(ESP)と福井関数()を評価し、ミクロな吸着機構を解析しました(図2)。
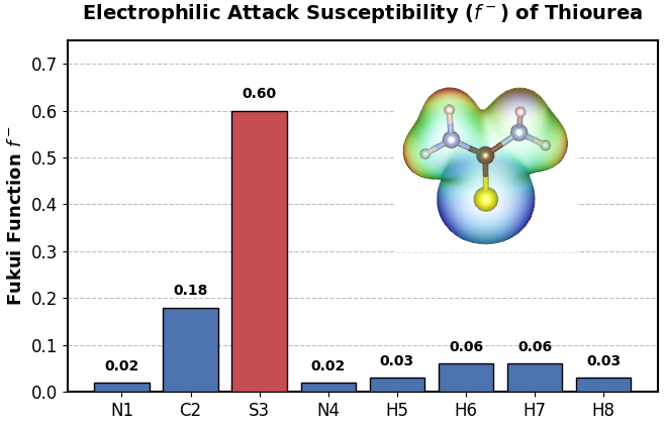
図2. チオ尿素の福井関数(\(f^-\))と静電ポテンシャル(ESP)マップ。ESPの可視化にはVESTA [3] を使用し、電子密度の等値面上にESPの値をカラーマッピングして作成しました。硫黄(S3)原子における局所的な高い反応性と、負の静電ポテンシャル(青色領域)の集中が確認できます。
解析の結果、分子内の硫黄(S)原子周辺に負の静電ポテンシャルが集中しており、かつ求電子的反応性(電子供与のしやすさ)の指標である福井関数 がS原子で特異的に高い値(0.60)を示しました。このことは、チオ尿素が窒素(N)ではなく硫黄(S)原子をアンカーとしてAu表面の活性サイトに選択的に配位・攻撃し、錯体化を引き起こすという微視的な反応メカニズムを裏付けています [4]。
まとめ#
第一原理計算ソフトウェアAdvance/PHASE(表面物理)とNWChem(錯体化学)の連携により、Auエッチング薬剤の開発における配位子設計と面選択性を評価するワークフローを構築しました。さらに、実用的なチオ尿素エッチャントに関する電子状態解析(静電ポテンシャルおよび福井関数の評価)を通じ、硫黄原子を介したミクロな吸着・錯体化メカニズムを明らかにしました。本手法を用いることで、新規薬剤のスクリーニングや、複雑な界面反応の熱力学的な理解を迅速に進めることが可能となります。
本解析の詳細や、研究への適用可能性に関するご相談はこちら
お問い合わせ参考文献#
- A. Davis and T. Tran, "Gold dissolution in iodide electrolytes", Hydrometallurgy 26, 163 (1991).
- E. Aprà et al., "NWChem: Past, present, and future", J. Chem. Phys. 152, 184102 (2020).
- K. Momma and F. Izumi, "VESTA: a three-dimensional visualization system for electronic and structural analysis", J. Appl. Crystallogr. 41, 653 (2008).
- G. K. Parker and G. A. Hope, "Spectroelectrochemical investigations of gold leaching in thiourea media", Miner. Eng. 21, 489 (2008).
関連ページ#
- 第一原理計算ソフトウェア Advance/PHASE
- 解析分野:ナノ・バイオ
- 産業分野:材料・化学