3D-RISM-SCFを用いたA型ゼオライトへの第一原理計算の応用#
溶媒は分子運動によって分子の位置が激しく入れ替わり一定の構造をとらないために、通常の第一原理計算が応用できません。ですが、Advance/PHASE では溶媒を取り扱う手法として 3D-RISM-SCF を導入し、溶媒への第一原理計算の適用を可能としました。ここではその理論の背景と、Advance/PHASE を用いたA型ゼオライトへのイオン吸着に関する第一原理計算の事例を紹介します。
3D-RISM-SCF法について#
第一原理計算における溶媒の取り扱い#
溶媒(液体)は分子の熱運動により一定の構造をとりません。これは、分子の揺らぎを統計力学的に取り扱う必要があり、通常の第一原理計算が適用できないことを示します。
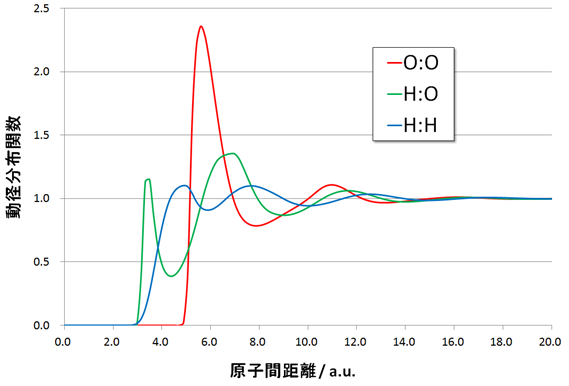
図1.水分子の動径分布 液体の水分子は一定の構造を持たない
よって、溶媒への第一原理計算の応用は、以下に示した手法・モデルのうちいずれかによって行う必要があります:
-
連続誘電体モデル
-
溶媒分子の明示的取り扱い
-
イオン系に特化した取り扱い
-
溶液論との連成
3D-RISM法#
3D-RISM 法とは、統計力学に基づく溶液論の実装方法の一つです。分子密度を用いたモデルであり、無限個の分子からなる系を取り扱い可能となります(計算には分子数密度関数と分子間相互作用関数を利用します)。分子一つ一つについてその座標や電荷分布を考慮する必要がなくなり、計算コストがかなり減少します。 3D-RISM 法の計算原理はこのようなものとなっています:
-
溶媒は、“分子” として取り扱う。 (分子内の構造は固定)
-
溶媒分子は、古典的な分子力場法にて近似する (電荷 + Lennard-Jones)
-
各原子を相互作用サイトとみなして、溶液論の積分方程式を解 → Reference Interaction Site Model (RISM)
-
原子間の相関が、動径分布関数として得られる
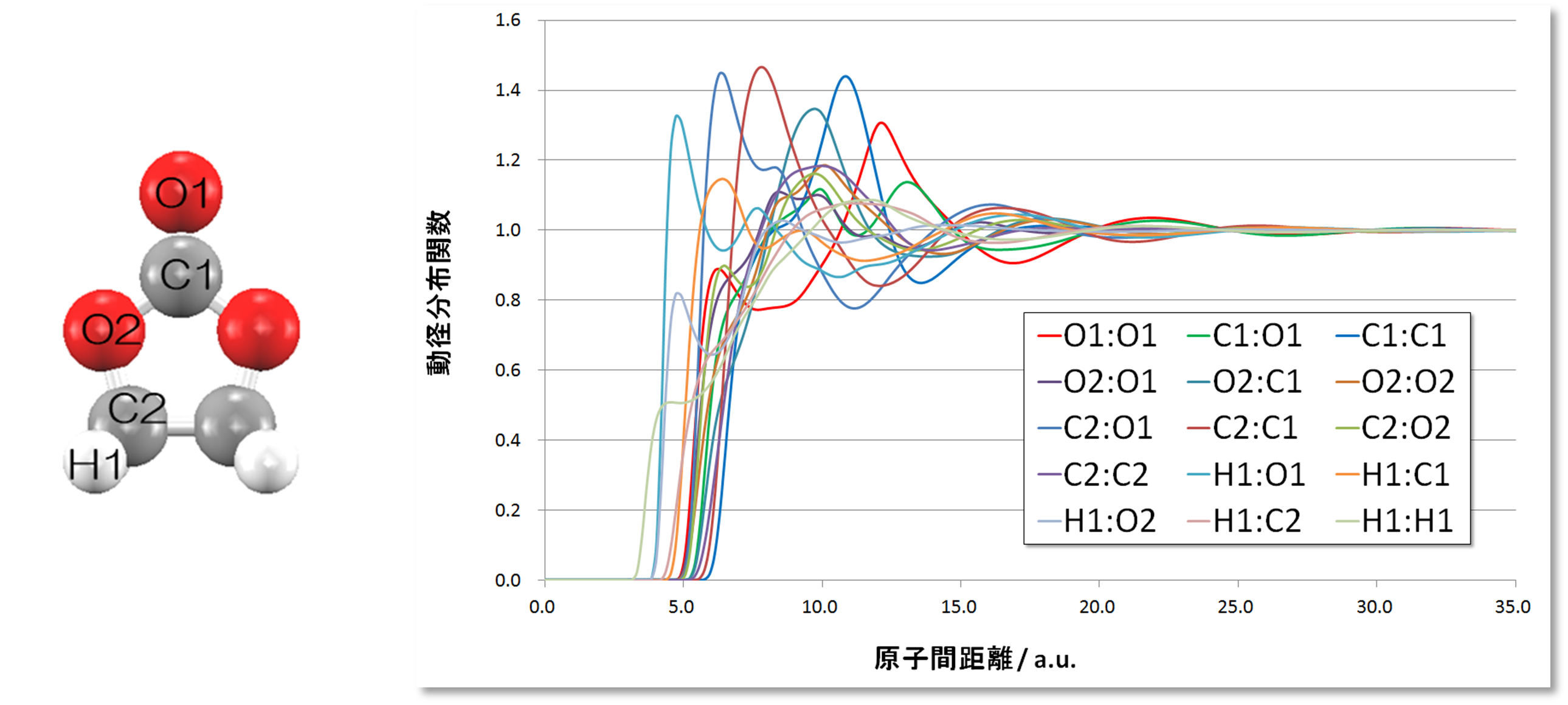
図2.エチレンカーボネートの動径分布
溶質-溶媒間の相関の計算については、予め計算された溶媒分子間の相関を利用して溶媒分子間の相関を考慮しながら、溶質-溶媒間の積分方程式の解を求めます。これによって各溶媒原子について、3次元(3D-)の分布関数が得られます。
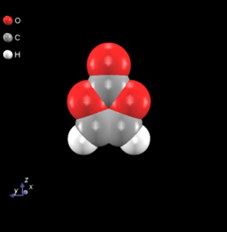
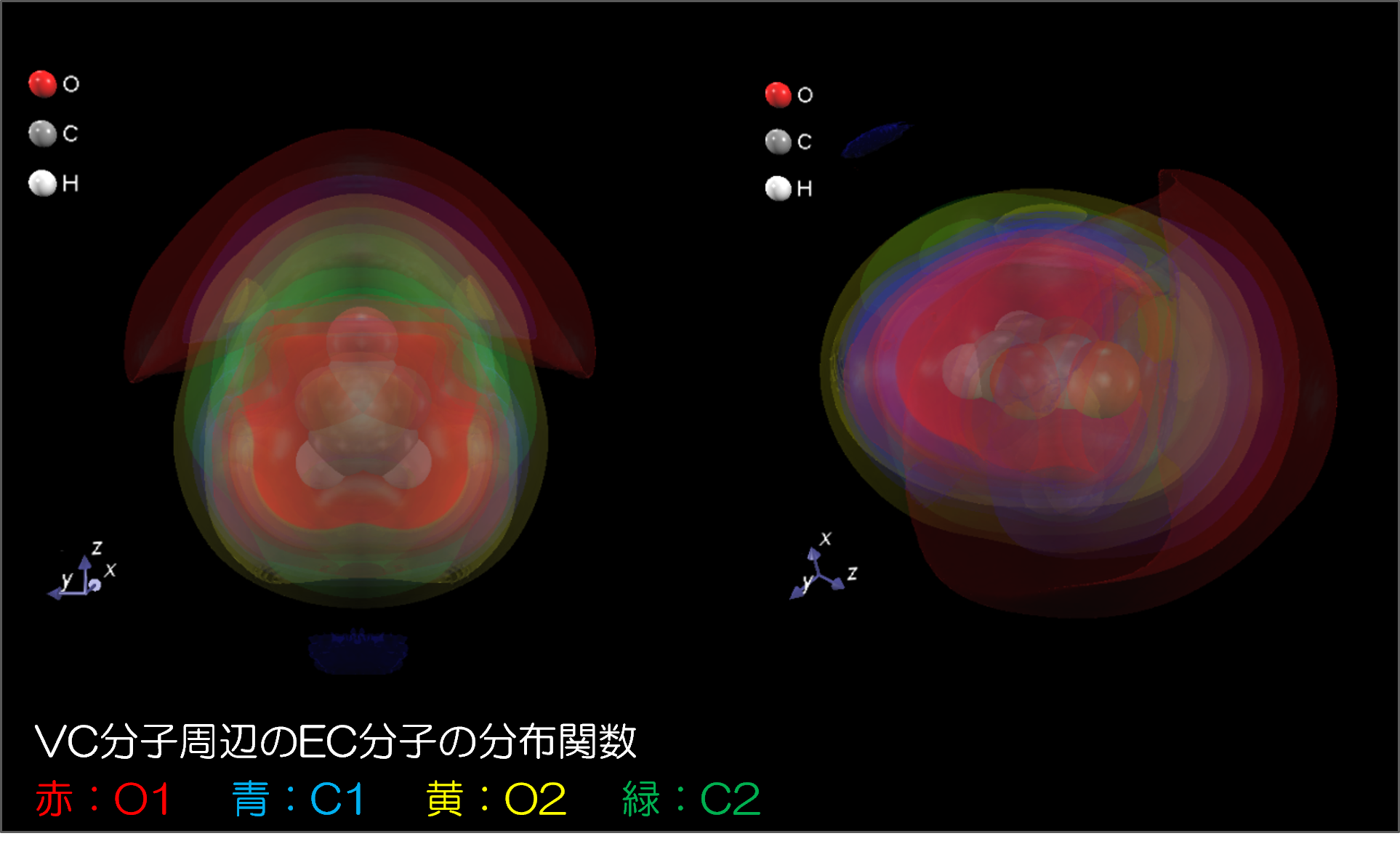
図3.ビニレンカーボネート(VC)分子周辺のEC分子の分布関数(赤:O1 青:C1 黄:O2 緑:C2)
3D-RISM-SCF法#
Advance/PHASE では以下の図4に示す手法により、第一原理計算と 3D-RISM 法の連成によって溶質に第一原理計算を適応して様々な材料計算を実施します (※ Lennard-Jones 力場のみ古典的に扱います)。まず、電子雲の作る静電ポテンシャルの下、溶媒分布を 3D-RISM で計算します。続いて溶媒の作る(溶媒和)ポテンシャルの下、電子状態をSCFで計算します。そして溶媒分布と電子状態を、自己無撞着になるまで最適化し、結果を出力します。
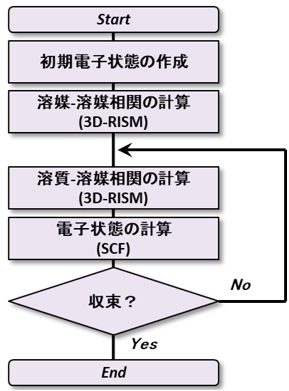
図4.計算のスキーム (3D-RISM-SCF法)
Advance/PHASE における 3D-RISM-SCF 法の実装は、予め計算された溶媒分子間の相関を考慮・利用し、溶質-溶媒間の積分方程式を解くという手法で行っています。これにより、各溶媒原子について、3次元( 3D- )の分布関数を得られます。この 3D-RISM-SCF 法は Advance/PHASE に商用ソフトでは世界で初めて実装されました。
計算可能なシミュレーション#
| 解析に用いる数値・関数 | 得られる結果 |
|---|---|
| 溶媒分布関数 | 溶媒分子・イオンの吸着サイトを特定 |
| 溶媒和自由エネルギー | 塩 ・極性分子の溶解に伴う、熱力学量を評価 |
| 溶媒効果を考慮した全エネルギー | 溶媒存在下での安定性評価、ポテンシャル曲面を計算 |
| 溶媒効果を考慮した構造最適化 | 溶液中の分子等の安定構造を探査 |
| 溶媒存在下でのバンド計算 | 電子状態に対する溶媒の影響を調査 |
計算手法の比較
※ 赤字 はより高性能であることを意味します。
| PCM, COSMO | Bluemoon, QM/MM | Poisson-Boltzmann | 3D-RISM-SCF | |
|---|---|---|---|---|
| 溶媒の取り扱い | 連続体 | 分子 | 連続体 (イオンに限る) | 分子 |
| 溶媒分子間の相互作用 | 無し | 有り | 無し | 有り |
| 溶媒分布の計算 | 不可 | 可 | 可 | 可 |
| 溶質系 | 分子 | 分子, 表面, 固体 | 分子, 表面, 固体 | 分子, 表面, 固体 |
| 統計力学的正当性 | 正 | ? (サンプリングに依存) | 正 | 正 |
| 計算コスト | 低 | 高 | 低 | 低 † |
†通常の実装方法では計算コストが高コスト ← Advance/PHASE は独自アルゴリズムで低コスト化を達成しました
計算事例:A型ゼオライト#
空孔内水分子数最適化シミュレーション#
まず、A 型ゼオライトの空孔内水分子数最適化シミュレーションを行いました。化学組成 Na12(H2O)27[Al12Si12O48] の A 型ゼオライトについて 3D-RISM-SCF 法で計算しました。負に帯電した骨格[Al12Si12O48]12- を溶質、 Na+イオンおよびH2O分子を溶媒として、3D-RISM-SCF 法を適用しました。使用した力場は水分子に対して SPC/Eで、それ以外に対して ClayFF としました。シミュレーションの結果、以下の図5に示すように、計算値と実験値がほぼ一致することがわかりました。
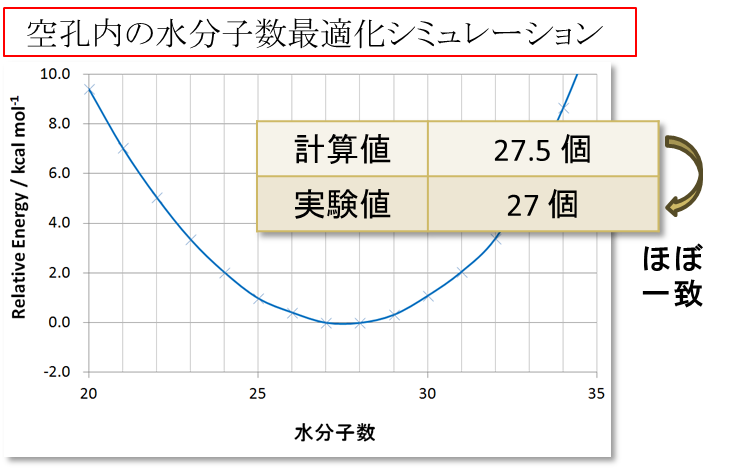
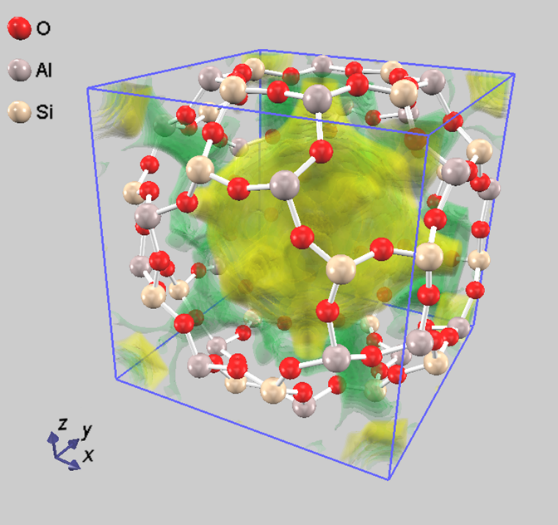
図5.A型ゼオライト内部での水分子の吸蔵
Na+イオンの吸着#
続いてNa+イオンの吸着に関する第一原理計算を実施しました。A 型ゼオライトは図6に示すように A、B、C 3つのカチオン吸着サイトが存在します。各サイトにはNa+が、以下の表に示す個数が吸着することが実験的に知られています†:
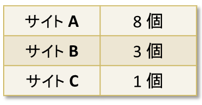
†R. Y. Yanagida, et al, J. Phys. Chem. 77, 805 (1973)
計算結果は次のようになりました。まず、 Na+イオンは サイト A に最も多く分布していることがわかりました。また、サイトBにおける分布関数の値はサイト A のそれより小さく、サイト C では極めて小さいこともわかりました。よって Advance/PHASE を用いることによって、 Na+イオン分布の実験データをシミュレーションで定性的に再現可能であるといえます。
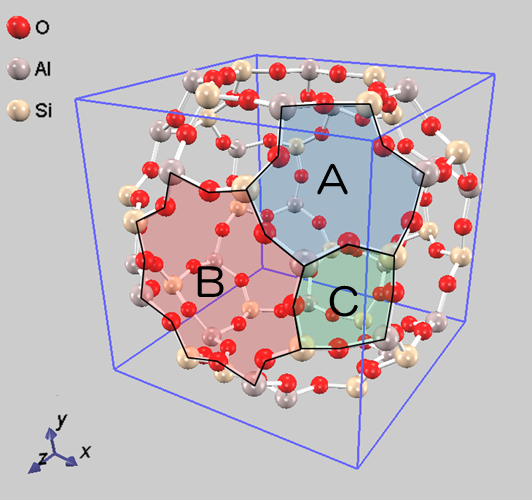
図6.A 型ゼオライト内のカチオン吸着サイトとNa+イオンの分布関数
イオン吸着におけるサイト選択性シミュレーション#
また 3D-RISM-SCF 計算による、イオン吸着におけるサイト選択性に関するシミュレーションも実施しました。この際 Na+の一部をK+に置換しました。各イオンの吸着サイト (実験値) は以下です:

図7は 3D-RISM-SCF 法により計算したNa+/K+イオンの分布関数を示しています(右側が拡大図です)。この結果は実験値と定性的に一致しています。
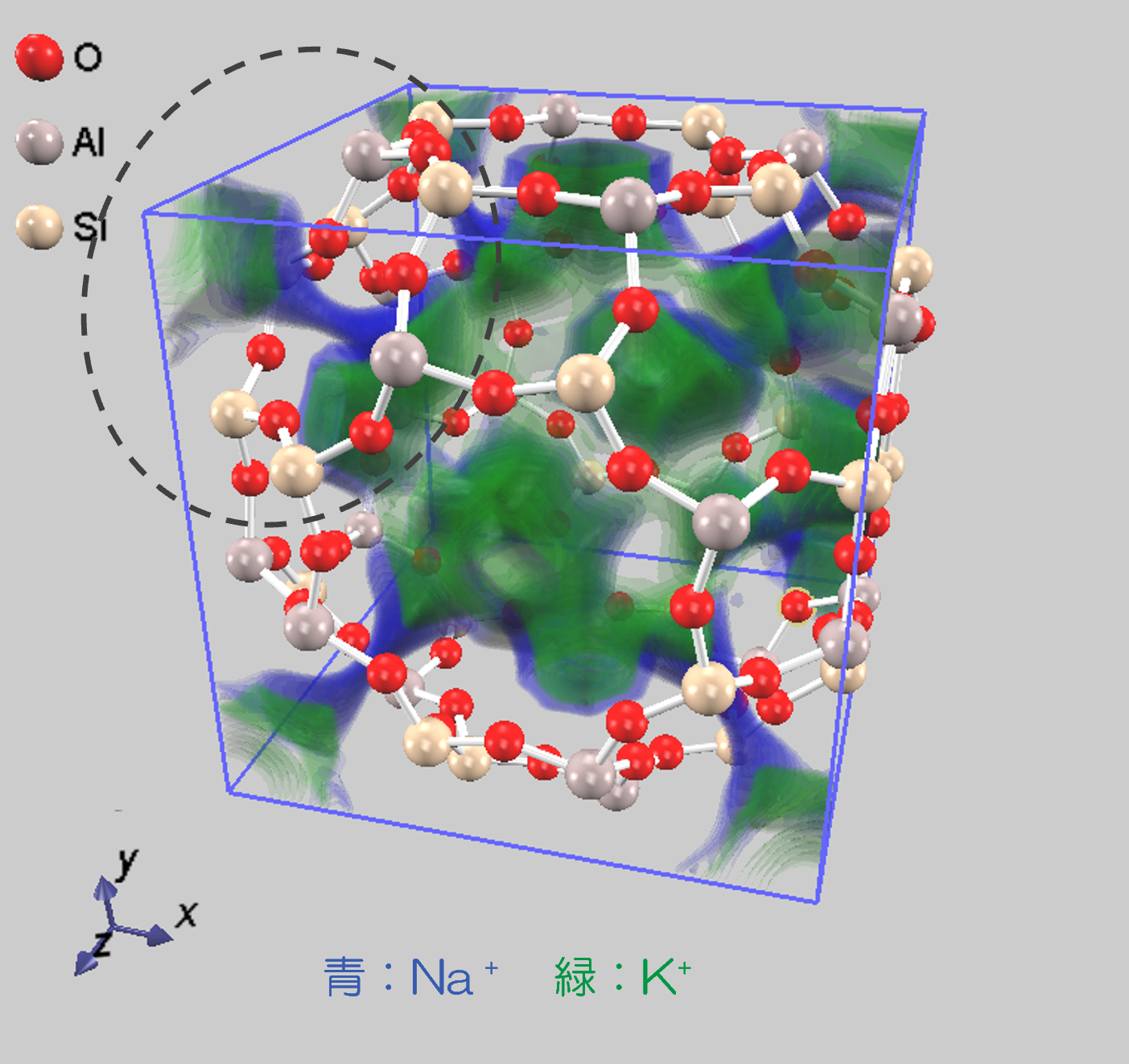
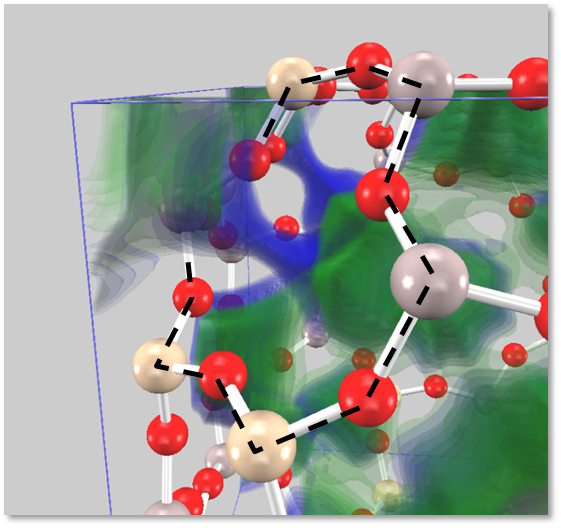
図7.3D-RISM-SCF 法により計算したNa+ / K+イオンの分布関数
電子状態への影響#
3D-RISM-SCF 計算で得られた電子密度を基に、状態密度 (DOS)を計算しました。図8より、溶媒に含まれるカチオンの種類の変化が、溶質 (ゼオライト骨格) の電子状態に影響を与えることがわかります。
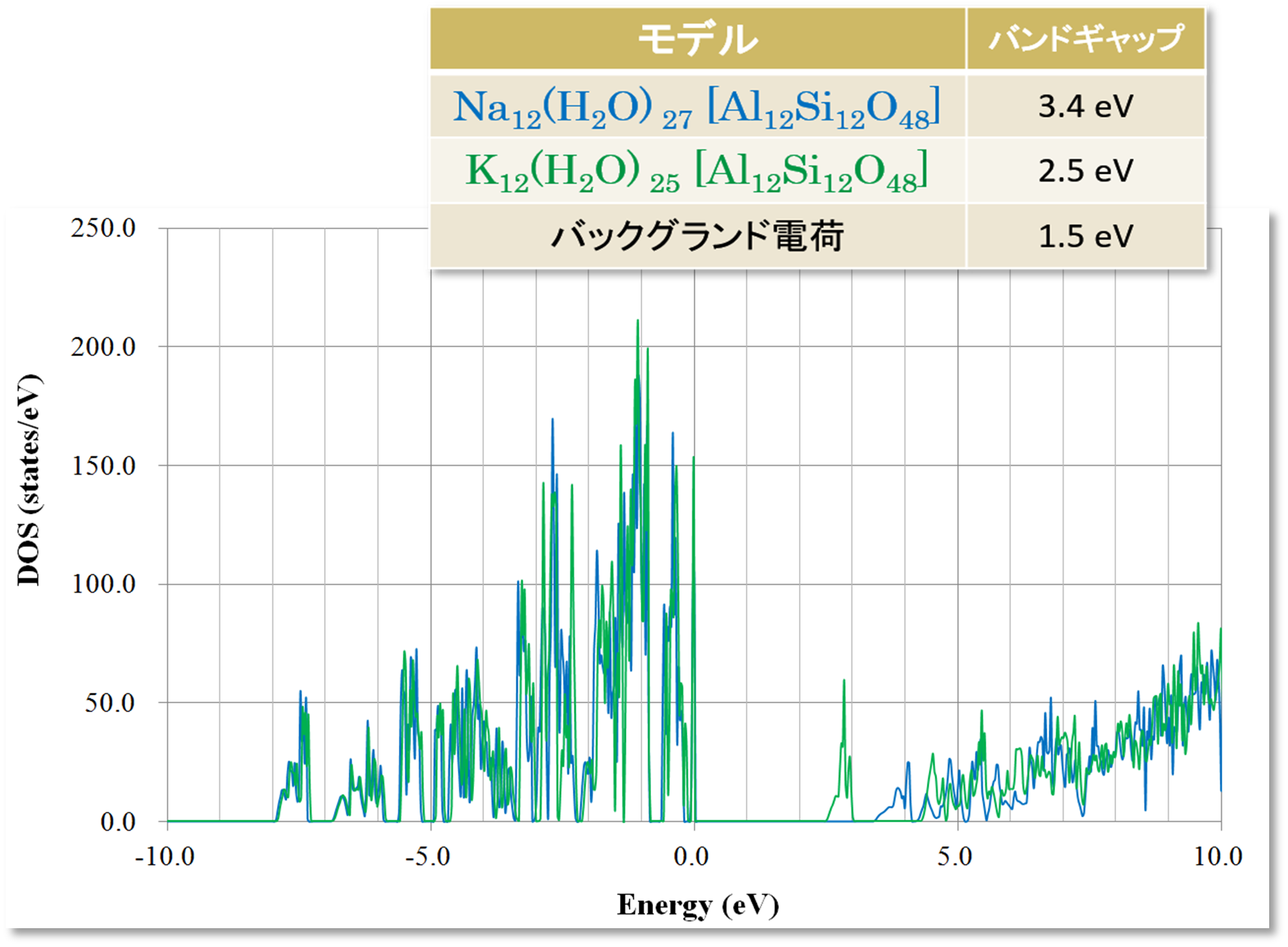
図8.A 型ゼオライトの状態密度 (DOS)とバンドギャップ
Na+イオンを含む場合、バンドギャップは 3.4 eV であり、K+イオンを含む場合、バンドギャップは 2.5 eV となります。
関連ページ#
- 第一原理計算ソフトウェア Advance/PHASE
- 解析分野:ナノ・バイオ
- 産業分野:材料・化学