Molecular Dynamics Simulation of Hydrogen adsorbed on Metallic Catalyst via Self-Learning Hybrid Monte Carlo method#
We created the Neural Network Potential (NNP) of the hydrogen adsorbate on the Pt3Ni alloy catalyst with self-learning hybrid Monte Carlo (SLHMC) method, and calculated the adsorption length and other data with Molecular Dynamics (MD) using NNP.
Create NNP via SLHMC method#
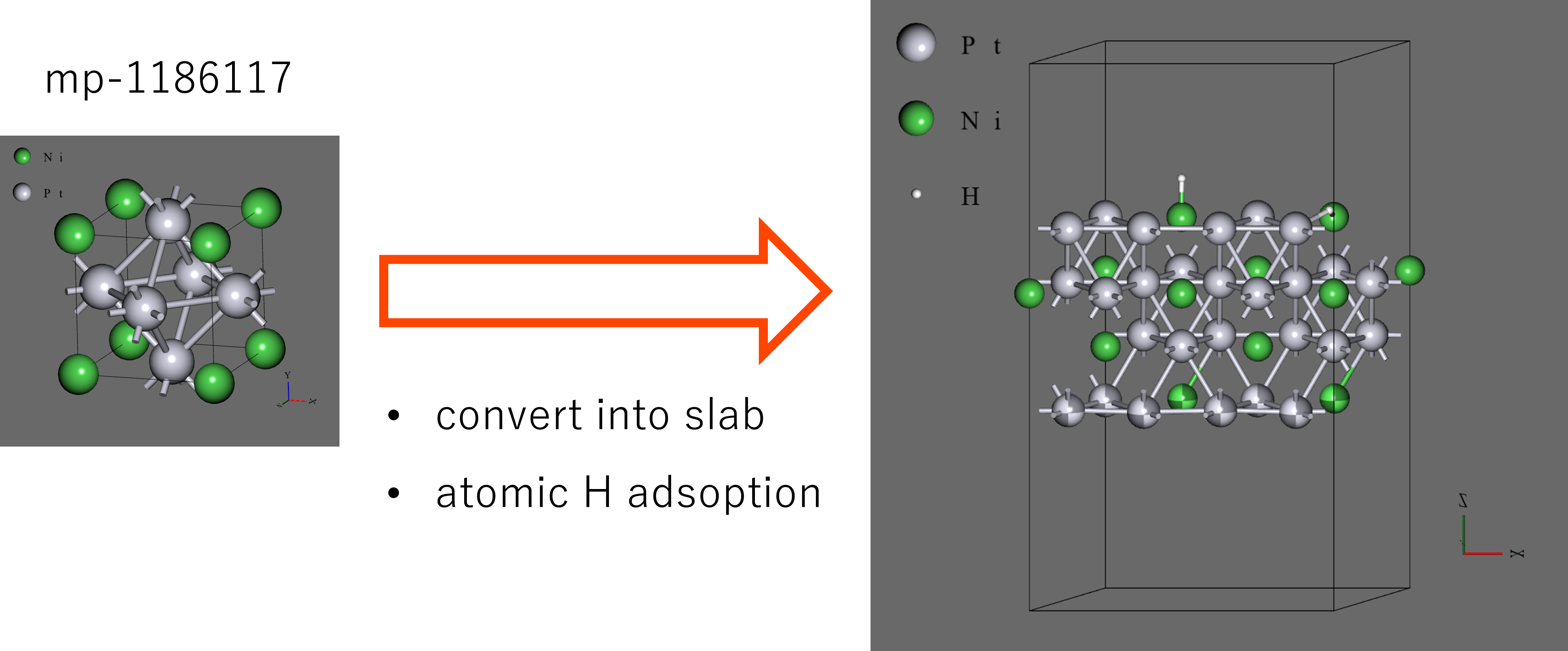
- For the initial structure, we obtained the bulk structure from Materials Project (mp-1186117), converted to a 4-atoms x 4-layers slab, replicated in the x-direction after structural optimization, and adsorbed 2 hydrogen atoms using Advance/NanoLabo features. The structure system has 34 atoms finally. To obtain the structural variation, only the 1 bottom layer of slab was fixed.
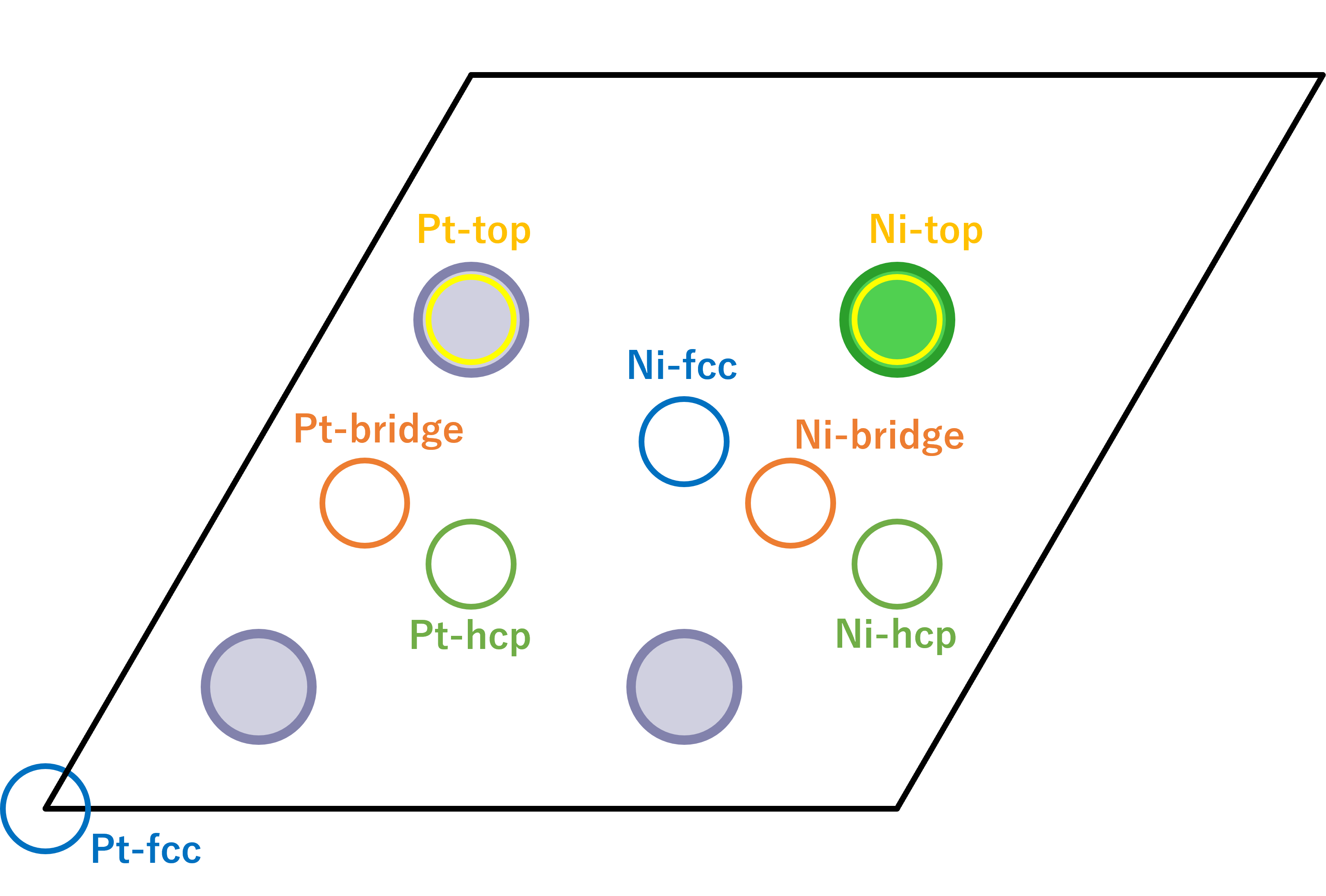
- The two adsorption sites were selected for the initial structure: top site on the Ni (Ni-top), and hcp site neighboring to Ni (Ni-hcp). To obtain the variation of length, the top site was put far, and the hcp site was put near the slab compared to real length.
- MD calculation performed with NPT ensemble. First-principles MD was done 200 steps for creation of initial NNP, and then NNP-MD was done 10 – 2000 steps (2.5 – 500.0 fs) per one SCF calculation. The temperature was settled at 400 K.
- The density functional theory (DFT) using ultrasoft pseudo potentials is used for SCF calculation. GGA-PBE functional was used as an exchange-correlation functional. The cut-off energy was 36.0 Ry, and k-points were 3 points in the y-direction. The spin polarization was not considered.
- The structure of Neural Network were 40-nodes x 2-layers, and twisted tanh was used as an activation function. 80 Chebyshev functions were used as symmetric functions, and the cut-off radius was settled at 6.5 Å.
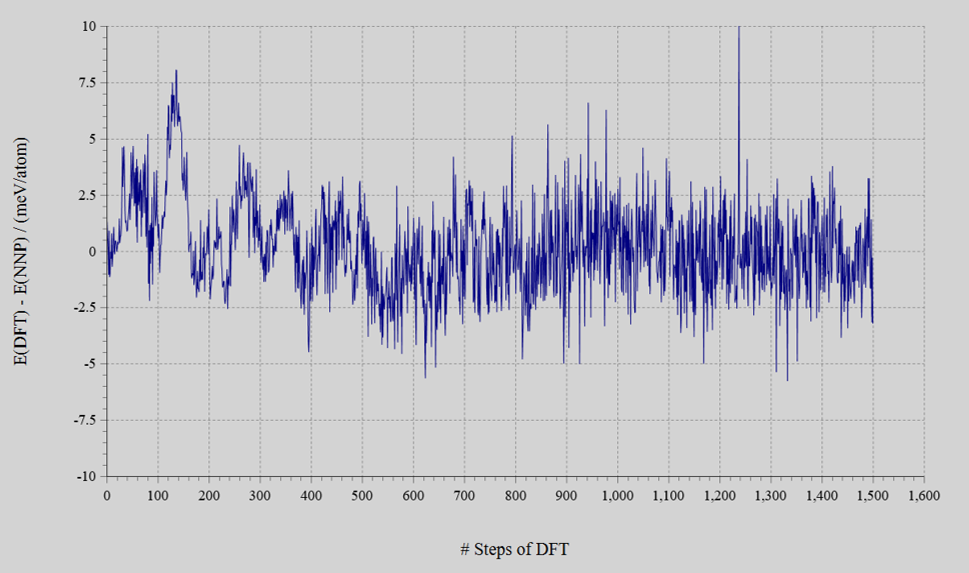
The creation of NNP completed in about 70 hours using Intel Xeon Gold 5218 (2 CPU, 32 cores).
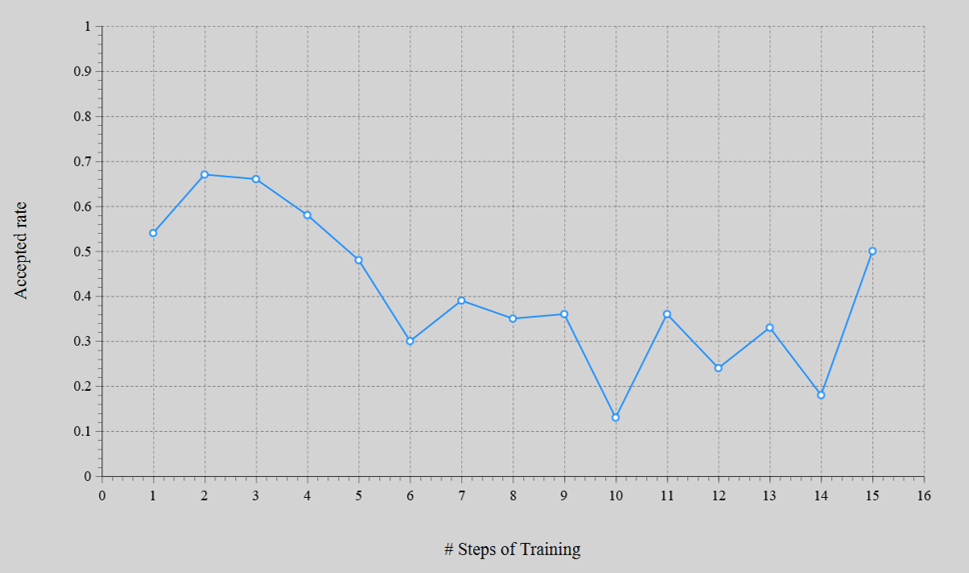
- The number of training data including the accepted and the rejected was 1700, finally.
- The energy error converged to MAE = 1.3 meV/atom. The acceptance rate on Monte Carlo method was in ranged 13 – 67%.
Calculation of Adsorption Length#
We optimize the structure using MD calculation with NNP (NNP-MD) and first-principles calculation with DFT, and compared the adsorption length and energies of each adsorption sites.
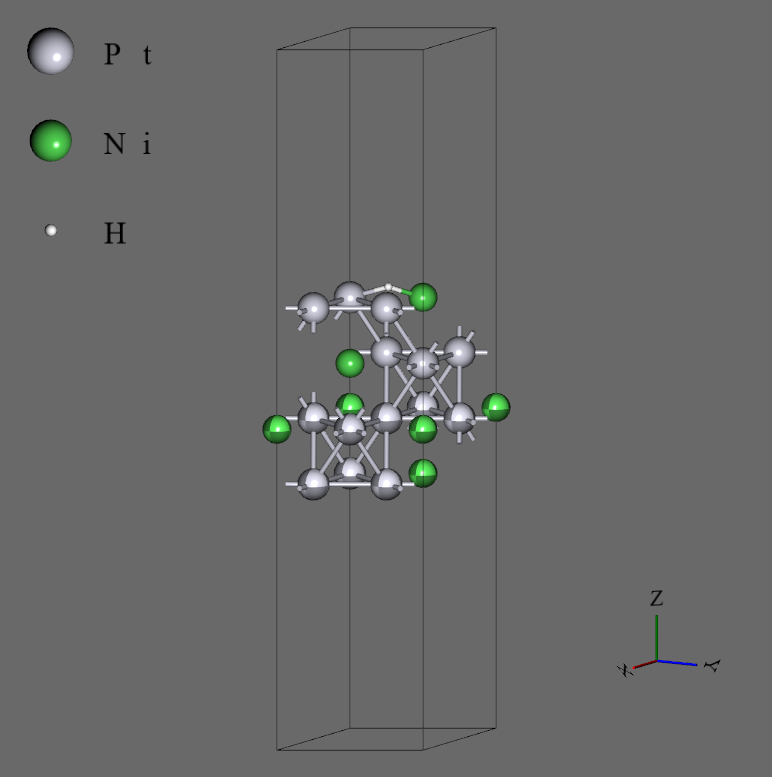
- For the structures, we use 4-atoms x 4-layers slab which was adsorbed a hydrogen atom on surface using Advance/NanoLabo features. The atomic coordinates of the 2 bottom layers were fixed.
- 8 adsorption sites are used: fcc, hcp, top, bridge sites neighboring/on the Ni, and these sites neighboring/on only Pt. The adsorption length of fcc and hcp sites were settled at 0 in default, so the initial structures was moved to 0.5 Å.
- To calculate adsorption energies, the structural optimization of a slab without adsorption and an atomic hydrogen was calculated too.
- DFT using ultrasoft pseudo potentials is used for SCF calculation. GGA-PBE functional was used as an exchange-correlation functional. The cut-off energy was 40.0 Ry. The k-points were 9 points total, 3 points each in the x- and y-directions. The spin polarization was colinear, and the initial polarizabilities of Ni, Pt and H are 0.2 μB, 0.0 μB, 0.0 μB respectively.
- The adsorption lengths were calculated as the difference between the z coordinates of hydrogen atoms and the average of the z coordinates of metal atoms in the top layer.
- The adsorption energies were calculated as the difference between the total energies of the slabs with adsorbate, and the sum of the total energies of the slab without adsorbates and the atomic hydrogen.
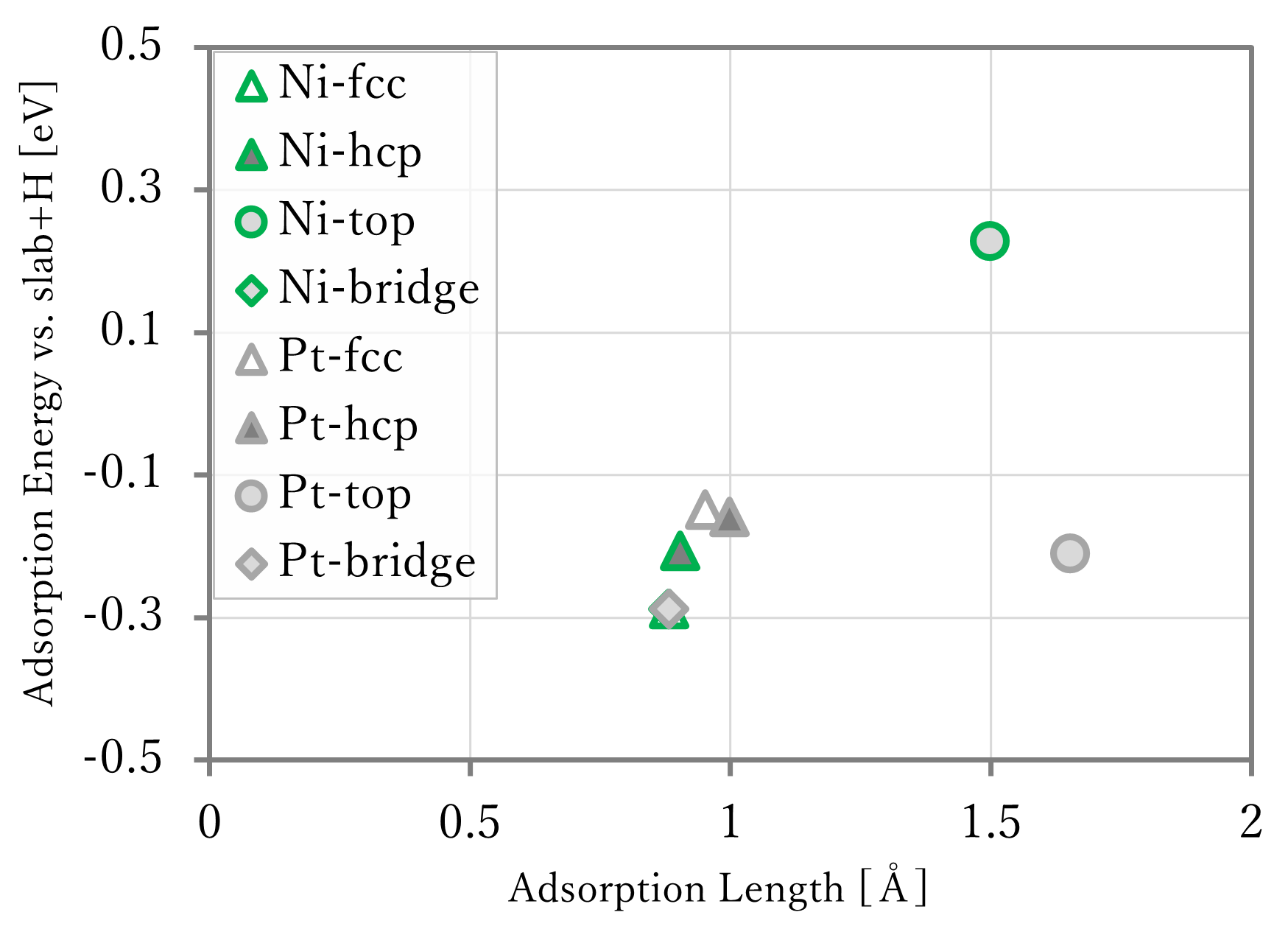
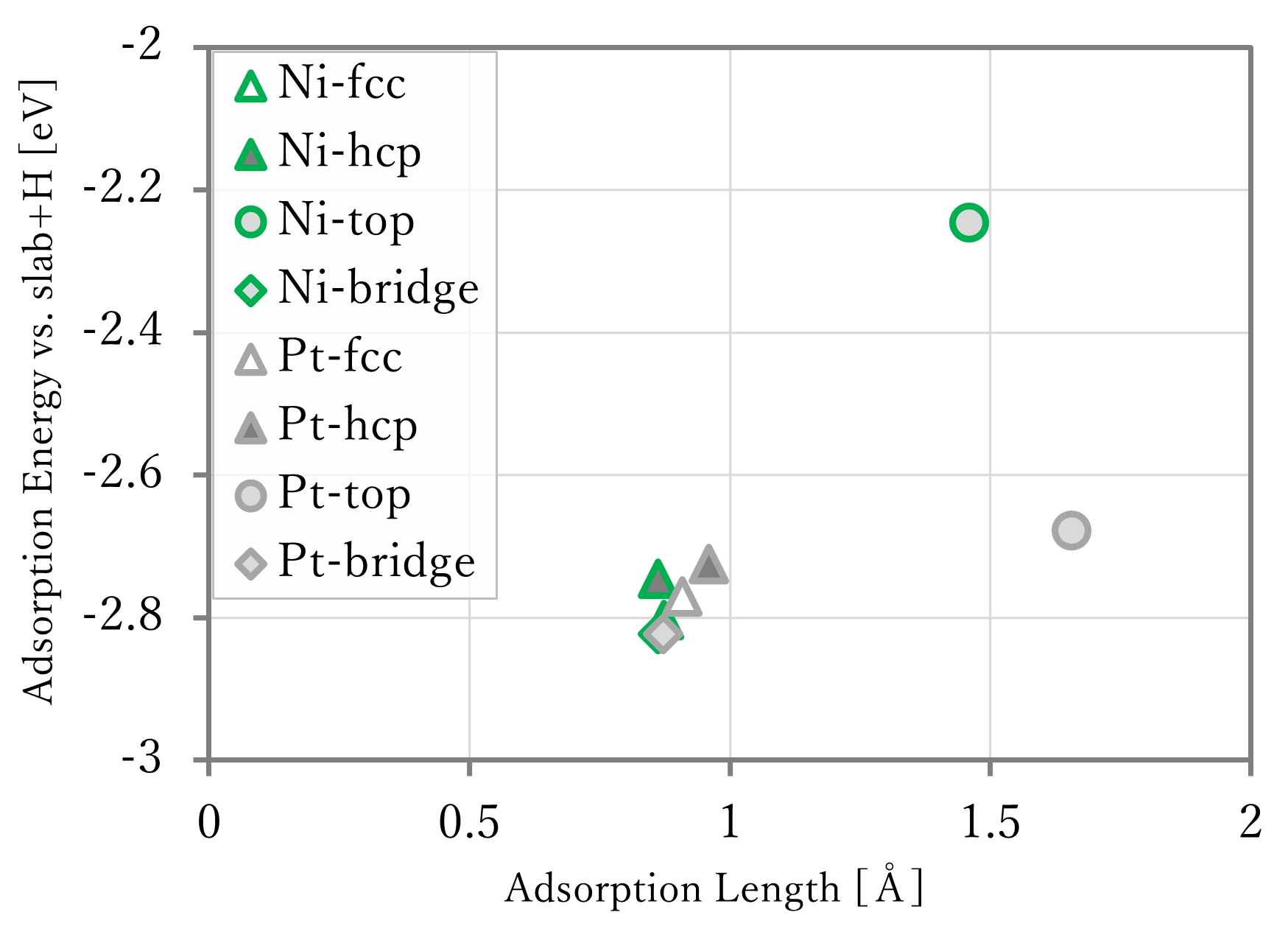
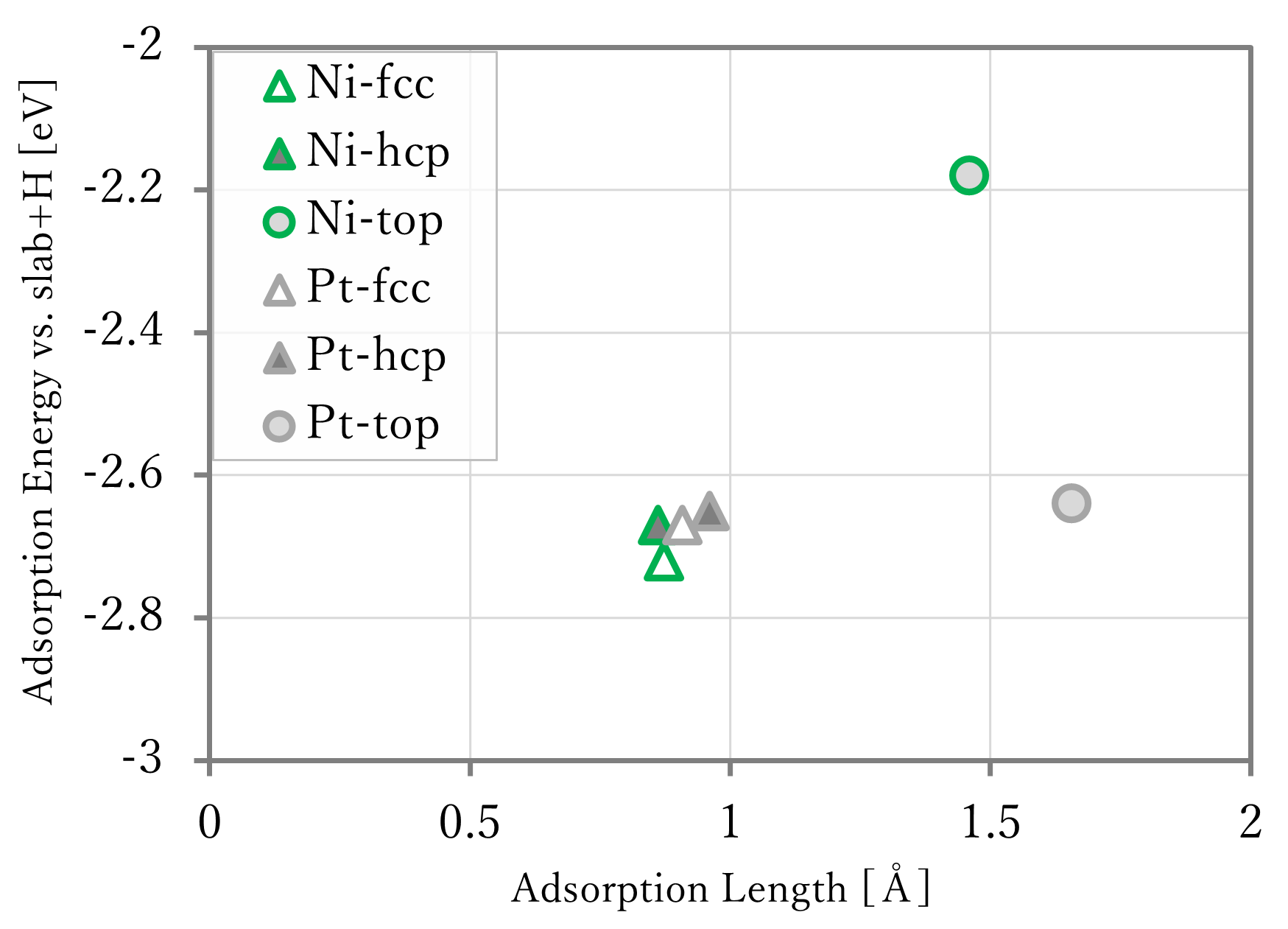
The results are shown in the below figure. In the right-below figure, the reference data[1] are shown too. However, because the lengths were not written in the reference, so that of the results using first-principles calculation was used in reference data.
- The adsorption lengths were well reproduced.
- The adsorption energies were generally reproduced, but the values of NNP-MD are different from that of first-principles calculation. This is probably caused by that the training data did not have data of slab without adsorbates, and that of atomic hydrogen.
Molecular Dynamics Simulation#
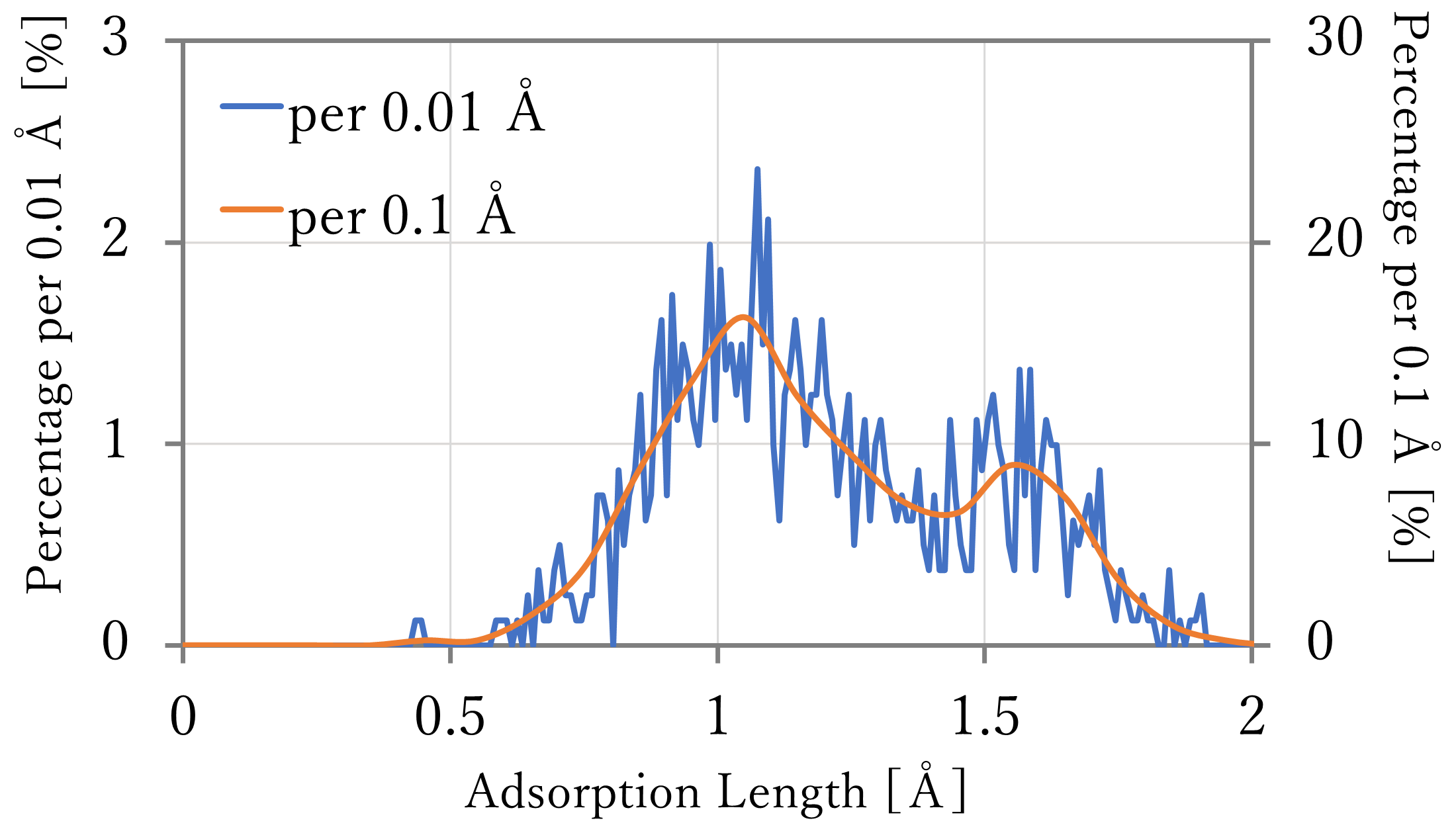
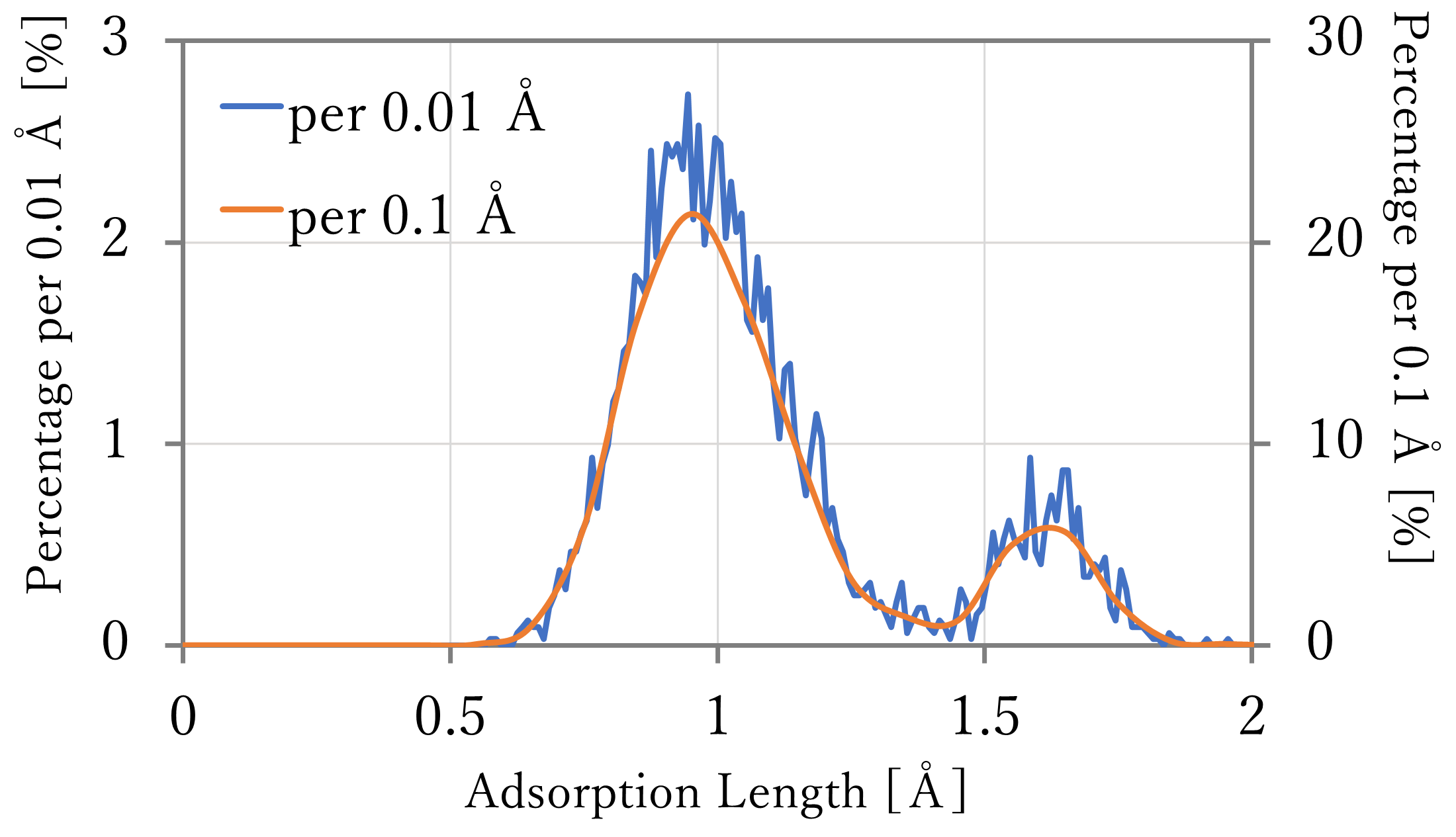
We calculated the distribution of the adsorption length of hydrogen atoms, using long-time NNP-MD simulations.
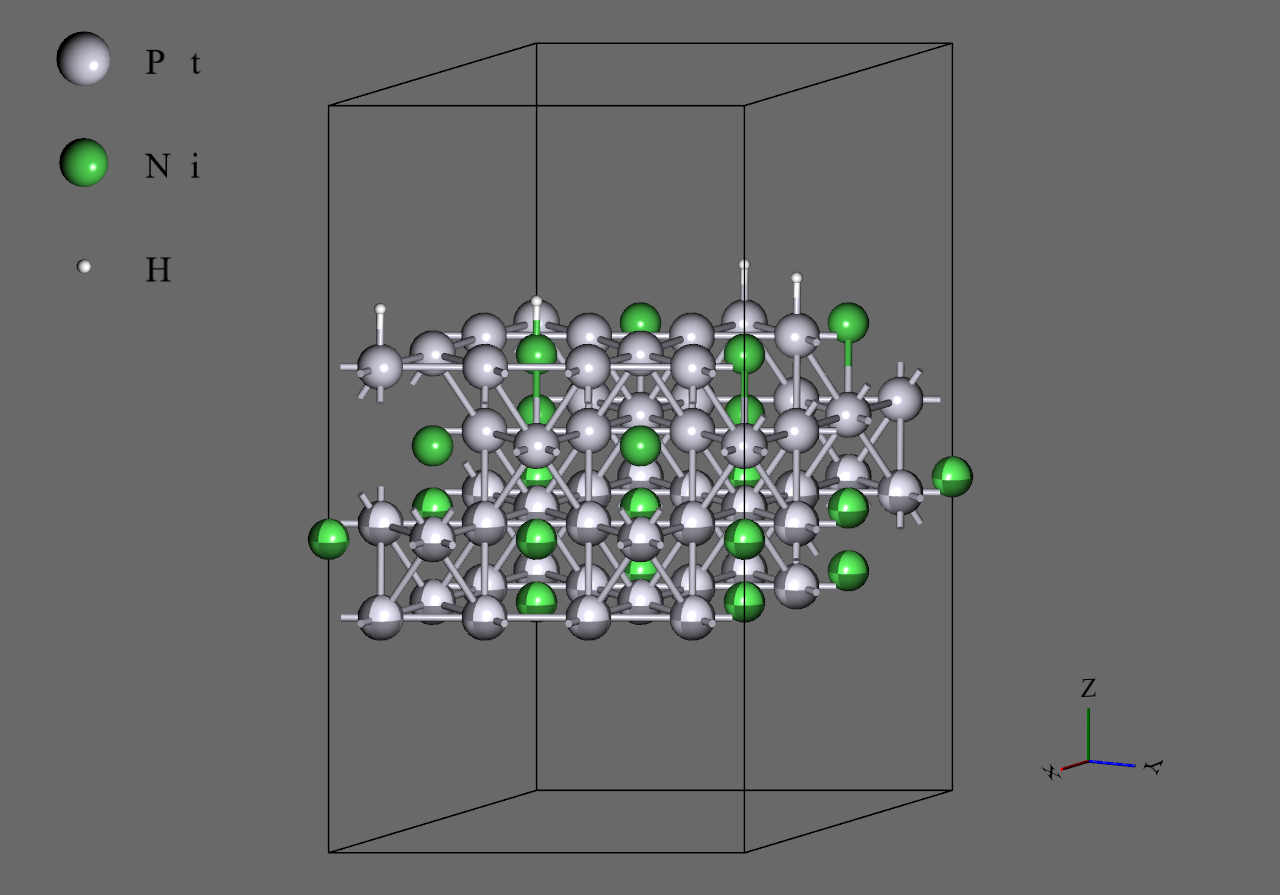
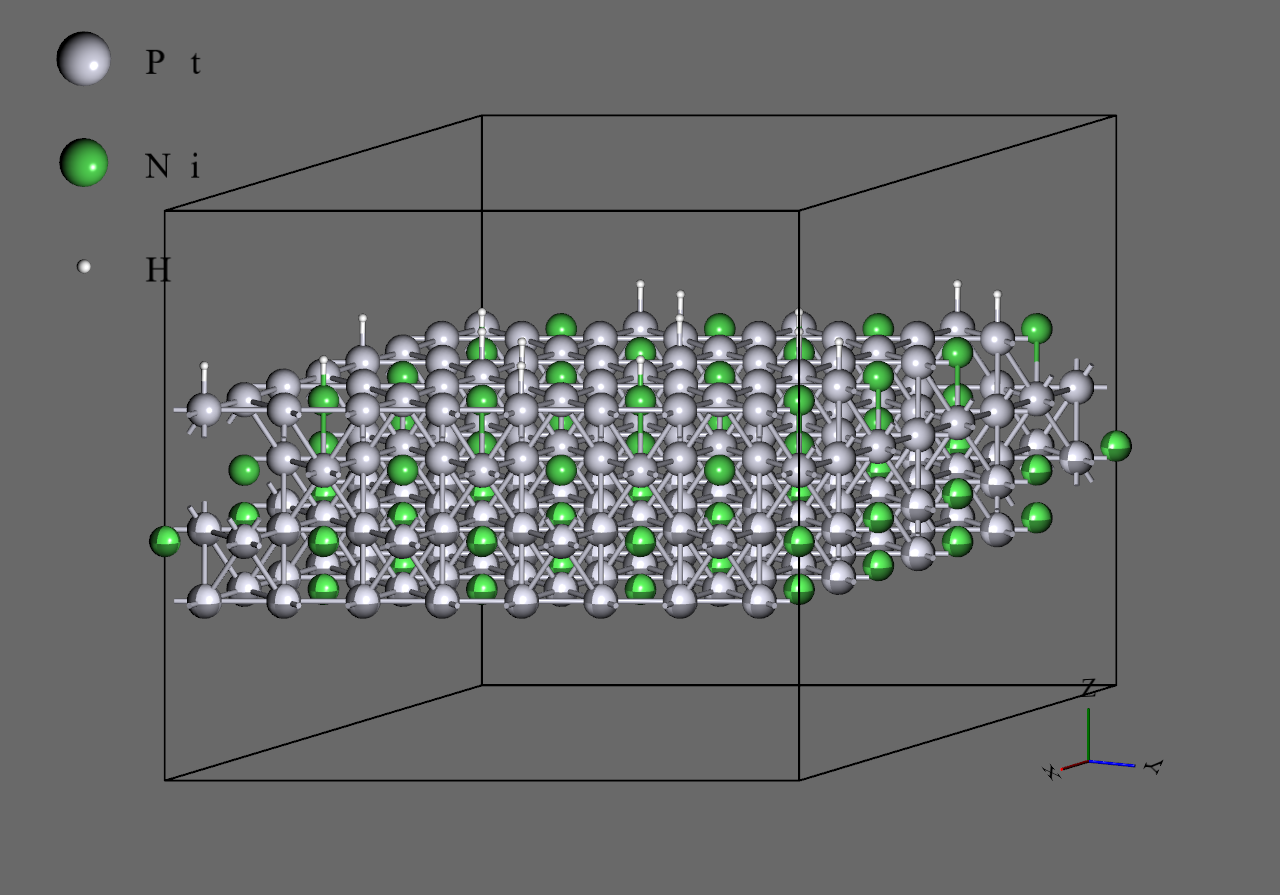
- For the structures, the 16-atoms x 4-layers slab with adsorbing 4 hydrogen atoms (68 atoms system) was used. Additionally, the 64-atoms x 4-layers slab was used too, which is the duplicated 16-atoms x 4-layers slab to x- and y-directions (272 atoms system).
Both were calculated with fixing the 2 bottom layers. - With NVT ensemble, the calculation was done 200,000 steps at Δt = 0.5 fs (total 100.0 ps). The temperature was settled at 300.0 K.
- The adsorption lengths were calculated as the different between the z coordinates of the hydrogen atoms and the average of the z coordinates of metal atoms in the top layer.
The atomic coordinates were outputted per 1,000 steps (0.5 ps), and the length was counted every 0.01 or 0.1 Å to calculate the percentage to the total.
- The calculation completed in about 20 and 55 minutes respectively. When the structural optimization written in the previous section was calculated using first-principles calculation, it took about 45 minutes. This shows that NNP-MD has low cost and high scalability.
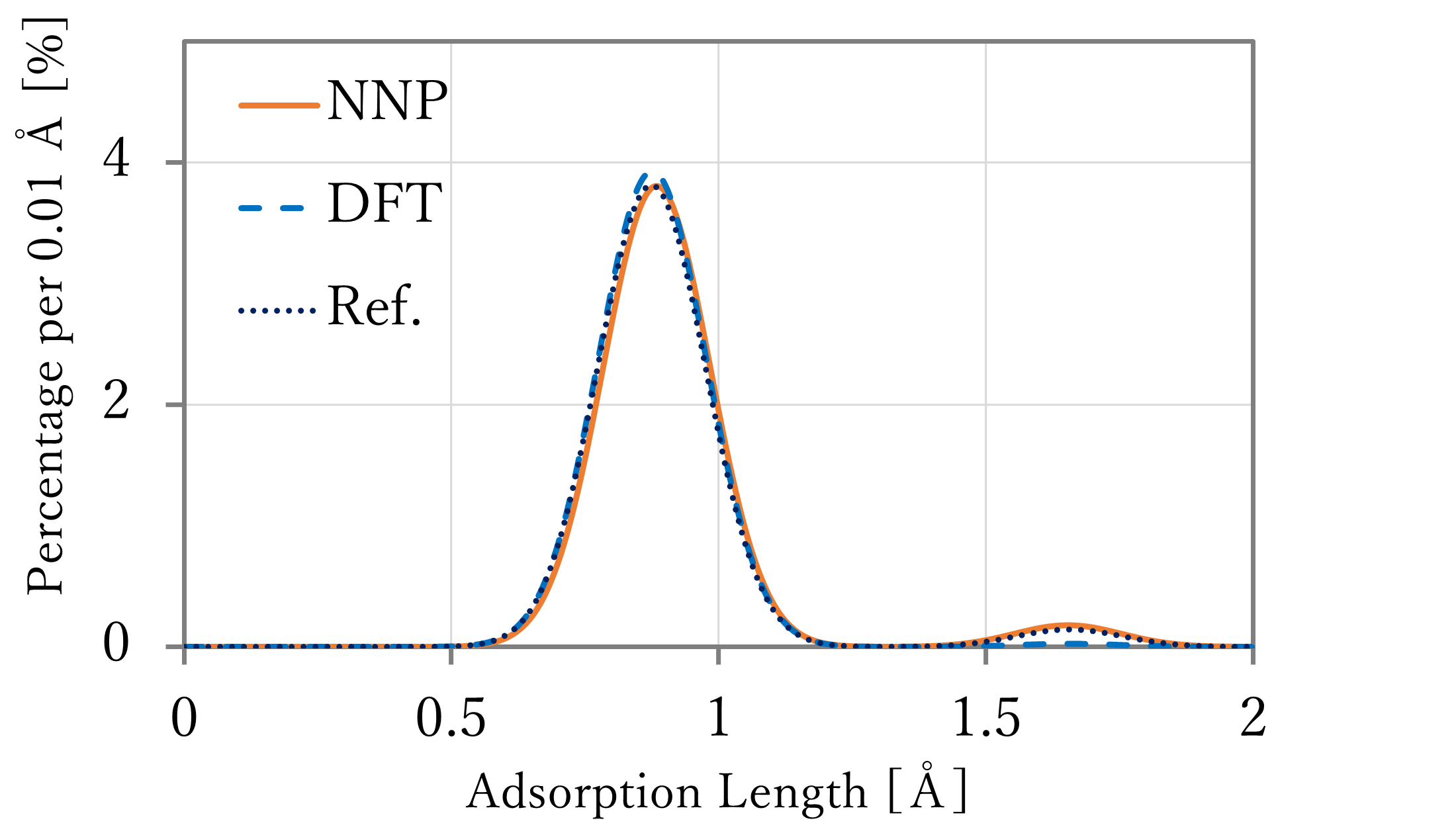
- The results are shown in the left-below figures. The blue and orange lines indicates the values counted per 0.01 and 0.1 Å respectively.
-
Additionally, the right-below figure shows the length distribution when the results of the adsorption length and energy calculations in the previous section are fitted to Boltzmann distribution and distributed by a normal distribution function with σ2 = 0.01.
Similar to the previous section, the length of result using first-principles calculation is used in the reference data. In addition, the results of the bridge sites of NNP-MD and first-principles calculation was excluded from the figure because all of these changed to hollow sites.
- As a result, the highest population were found at around 0.9 Å or 1.1 Å respectively, and in both results, some atoms existed at around 1.6 Å which seems the top sites. Comparing with the reference, the length of the hollow sites and around them were far a little, but the relation of the quantity was well reproduced, especially in the 64-atoms x 4-layers system.
- Comparing with the distribution fitted to Boltzmann distribution, there are a lot of structure between the hollow sites and top sites in the result of NNP-MD calculation. It is thought this difference caused because the calculation in the previous section was done at only sites which have high symmetry and stability. By using NNP-MD calculation, more close-to-reality results which include various structures can be get, with being based on the accurate prediction of energy.
16-atoms x 4-layers + 4H
64-atoms x 4-layers + 16H
Structural Optimization
関連ページ#
- ニューラルネットワーク分子動力学システム Advance/NeuralMD
- 解析分野:ナノ・バイオ
- 産業分野:材料・化学
- Advance/NeuralMD Product Information
- Advance/NeuralMD Documentation
-
S.Zhang et al., Topics in Catalysis, 2020, 63, 714–727 ↩