ニューラルネットワーク力場を使ったAlの分子動力学シミュレーション#
分子動力学シミュレーションでは、与えられた原子構造からある関数を使ってポテンシャルエネルギーを計算します。この関数は「力場」と呼ばれ、通常、経験的に決められた関数形を使います。一方、ニューラルネットワーク力場では経験的な知識を使わずに、学習によって0から力場を構成します。
本事例では、温度変化によるAlの状態変化のシミュレーションを行います。金属に適する力場としてはEAM(Embedded Atom Method、原子挿入法)力場が知られていますが、今回は既存の力場を使わずに、Advance/NeuralMDを使ってニューラルネットワーク力場を作り、現象の再現を試みます。
教師データの作成とニューラルネットワークの学習#
ニューラルネットワーク力場では、系の中の1つひとつの原子について、「近傍の原子構造」から「その原子のエネルギー、およびその原子に働く力」を求めます。そのためニューラルネットワークの学習に使う教師データとして、本番の計算で現れ得る「近傍の原子構造」を万遍なく用意する必要があります。

今回は、まず108個のAl原子からなる面心立方格子のモデルを元に、EAM力場を使った分子動力学シミュレーションで500 Kから3000 Kに昇温する過程を計算しました。これにより、この温度範囲で現れ得る原子構造が生成できます。次に、生成した構造の中から20個を抽出し、それぞれからAdvance/NeuralMDの機能を使ってランダムに変位させた構造を10個ずつ、計200個生成しました。
これらの200個の構造に対し、Quantum ESPRESSOを使って第一原理電子状態計算を実行し、エネルギーと力を計算しました。計算する系は200個ですが、系の中の1つひとつの原子から教師データ(「近傍の原子構造」と「その原子のエネルギー、およびその原子に働く力」との組み合わせ)が得られるので、データの数は108×200=21600個になります。
この教師データを使って、Advance/NeuralMDによりニューラルネットワークの学習を行いました。ニューラルネットワークの大きさはユーザーが指定することもできますが、今回はデフォルトの512ノード・2層としました。学習の結果、教師データに対する誤差(RMS)はエネルギー:0.4 eV、力:0.2 eV/Å程度まで最適化することができました。
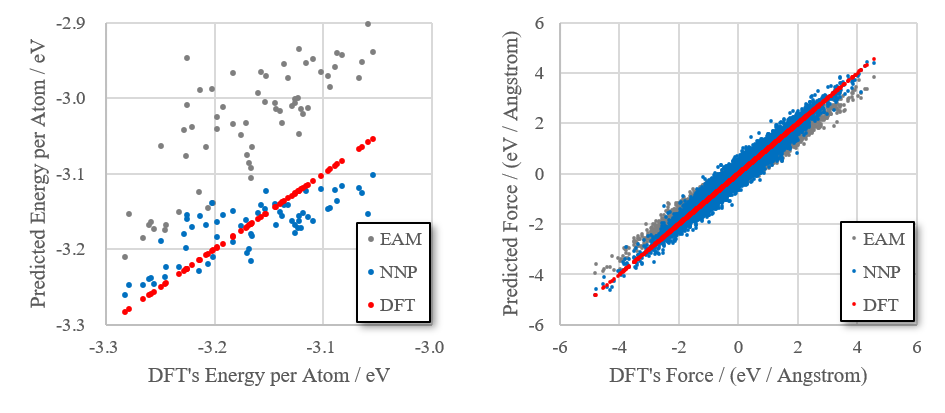
作成したニューラルネットワーク力場と、EAM力場を比較しました。最初の分子動力学シミュレーション(500 Kから3000 Kに昇温する過程)から1200~2000 Kの構造64個を抽出し、第一原理計算(DFT)、ニューラルネットワーク力場(NNP)、EAM力場を使ってそれぞれポテンシャルエネルギーと原子に働く力を求め、プロットしました。DFTとの差が小さいほど、良い力場ということになります。エネルギー、力ともNNPの方がEAMよりも良くDFTを再現していることが分かります。
分子動力学シミュレーションの実行結果#

作成したニューラルネットワーク力場を使い、500個のAl原子からなる面心立方格子のモデルでNVTアンサンブルの分子動力学シミュレーションをLAMMPSで実行しました。25 psで1 Kから2000 Kに昇温、続けて25 psの間2000 Kを維持し、溶融状態にします。その後、450 psかけて2000 Kから1 Kに冷却しました。
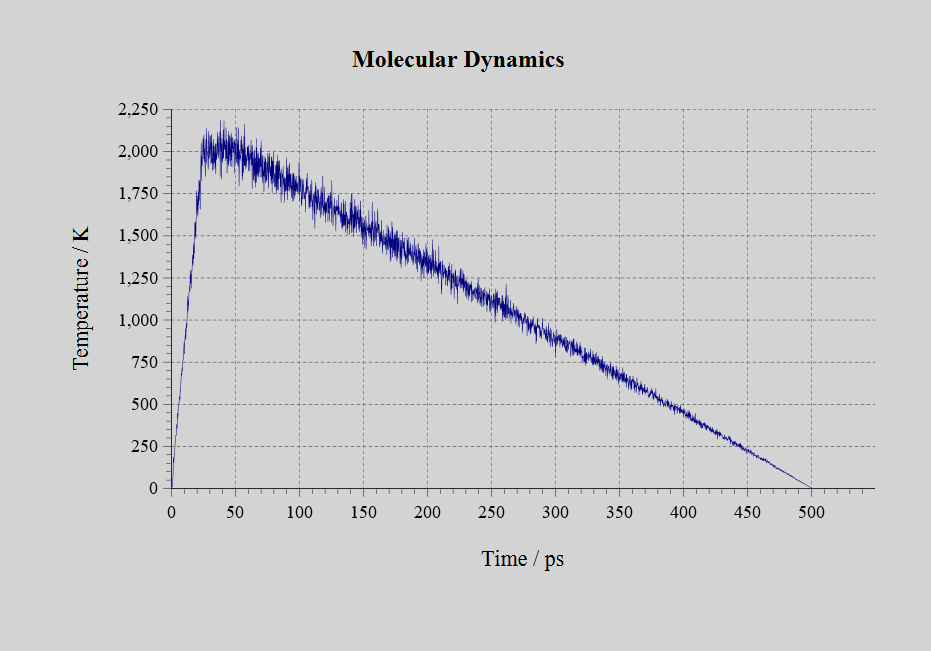
シミュレーションの結果を動画で見てみると、冷却の過程で結晶構造が現れる様子が分かります。
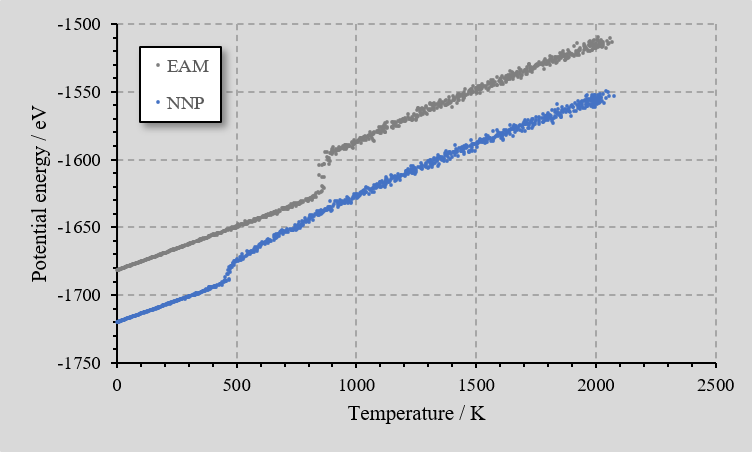
比較のためEAM力場でも同じ分子動力学シミュレーションを行い、冷却時の温度とポテンシャルエネルギーの関係をプロットしました。相転移に対応する不連続な個所があることが分かります。実際のAlの融点とは一致しませんが、温度変化に伴う状態変化の振る舞いを定性的に再現できています。