Molecular Dynamics Simulation of Al Atoms Using a Neural Network Force Field#
In molecular dynamics simulation, potential energy is calculated from a given atomic structure using a certain function. This function is called "force field", and usually is an empirically determined functional form. On the other hand, a neural network force field does not use empirical knowledge, but constructs a force field from zero through learning.
In this case study, a simulation of Al state change due to temperature change was performed. While EAM (Embedded Atom Method) force field is known as a force field suited for metal, this time an attempt to reproduce the phenomenon was made by creating a neural network force field using Advance/NeuralMD without conventional force fields.
Creating Training Data and Learning a Neural Network#
In a neural network force field, for each atom in the system, "energy of that atom and force acting on that atom" are determined from "adjacent atomic structure", which requires uniform preparation of "adjacent atomic structure" that potentially appears in the main calculation as training data for learning a neural network.

In this study, first the process of heating up from 500 K to 3000 K was calculated by molecular dynamics simulation using EAM force field based on a face-centered cubic lattice model consisting of 108 Al atoms. This allowed the generation of atomic structures that could appear in this temperature range. Next, 20 of the generated structures were extracted and 10 randomly displaced structures from each were generated using the Advance/NeuralMD functionality, for a total of 200 structures.
First-principles electronic structure calculations were performed on these 200 structures using Quantum ESPRESSO to calculate energy and force. Although there were 200 systems to calculate, the number of data was 108×200 = 21,600, since each atom in the system provided training data (a combination of "adjacent atomic structure" and "energy of that atom and force acting on that atom").
A neural network was trained by Advance/NeuralMD using this training data. While the size of the neural network can be specified by the user, in this time, the default size of 512 nodes and 2 layers was used. As a result of the training, the error (RMS) with respect to the training data was optimized to about energy: 0.4 eV and force: 0.2 eV/Å.
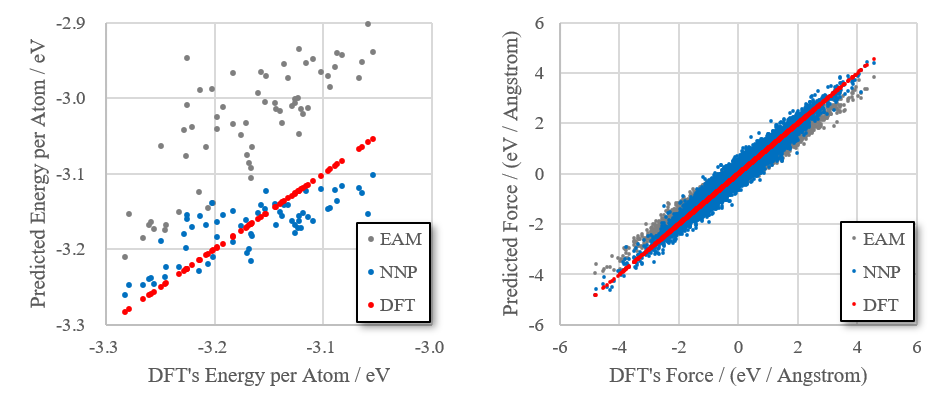
The created neural network force field was compared to the EAM force field. From the first molecular dynamics simulation (the process of heating up from 500 K to 3000 K), we extracted 64 structures between 1200 and 2000 K. Potential energy and force acting on the atoms were calculated using first-principles calculations (DFT), neural network force field (NNP), and EAM force field respectively, which were plotted. The smaller the difference from DFT was, the better the force field was. It could be seen that NNP force field reproduces the DFT better than EAM force field in terms of both energy and force.
Result of molecular dynamics simulation#

Molecular dynamics simulation of the NVT ensemble were performed in LAMMPS on a face-centered cubic lattice model consisting of 500 Al atoms using the created neural network force field. The temperature was raised in 25 ps from 1 K to 2000 K, and maintained 2000 K for 25 ps for the molten state. Then, It was cooled from 2000 K to 1 K in 450 ps.
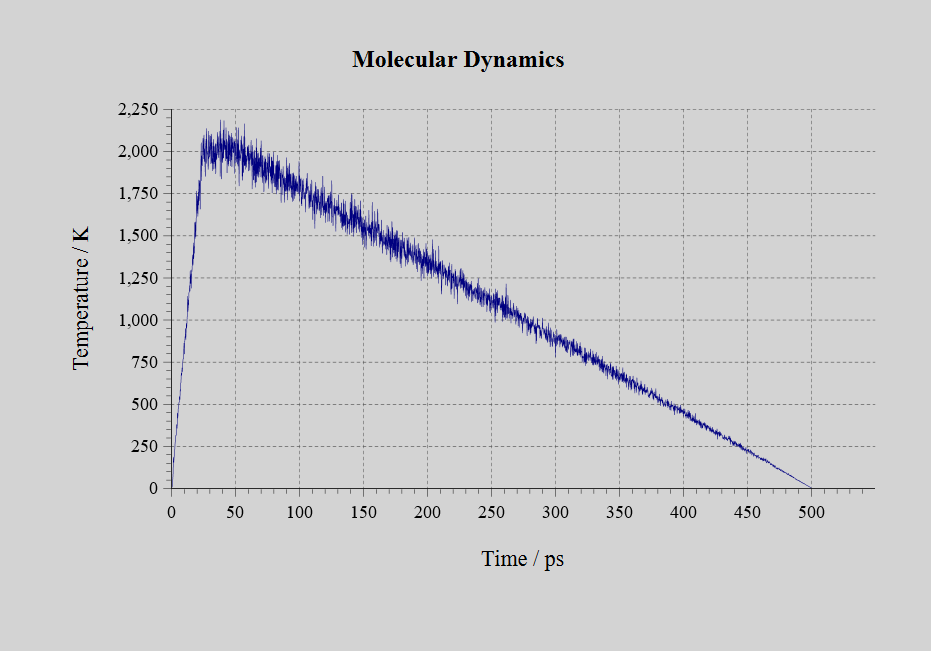
The animation of the simulation results shows how the crystal structure appears during the cooling process.
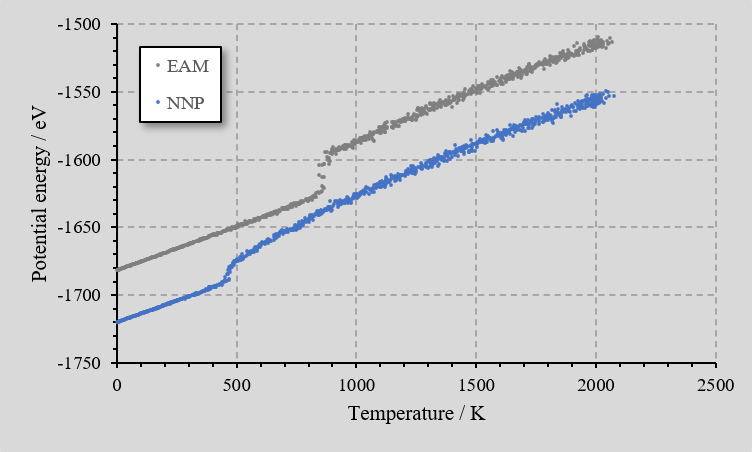
For comparison, the same molecular dynamics simulation was performed for the EAM force field, and the relationship between temperature and potential energy during cooling was plotted. It could be seen that there were discontinuities corresponding to the phase transition. Although it did not match the actual melting point of Al, it qualitatively reproduced the behavior of state change with temperature change.