白金(111)表面における一酸化炭素分子の吸着構造の密度汎関数理論による計算#
ナノ材料統合GUI Advance/NanoLaboを用いた、白金(Pt)(111)表面に一酸化炭素(CO)分子を吸着させた構造についての密度汎関数理論(DFT)による解析事例を紹介します。
紹介する事例は、モデルの作成からジョブの投入・結果の解析に至る一連のプロセスをすべてAdvance/NanoLabo上で行うことができます。
白金単結晶の計算#
最初のステップとして、白金単結晶のバルク構造の計算を行います。構造最適化の結果、格子定数が4.02Åの構造が得られました。実験値が3.92Å1であり、実験値と計算値の相対誤差は2.5%となっています。

|

|
|
構造最適化計算のエネルギー変化 |
最適化した白金のバルク構造 |
Pt(111)表面の計算#
最適化を行ったバルク構造から(111)表面が露出するように切り出した4層のスラブモデル(Pt(111))を作成します。表面の法線方向に十分な厚さの真空層を設定することで、3次元の周期境界条件のもとで表面モデルの計算を行うことができます。
本事例では下の2層の原子を固定してスラブモデルの構造最適化を行いました。

|

|

|
露出させる表面の面指数の設定 |
スラブモデル(Pt(111))の作成 |
固定する原子の設定画面 |

|
||
構造最適化後のスラブモデル |
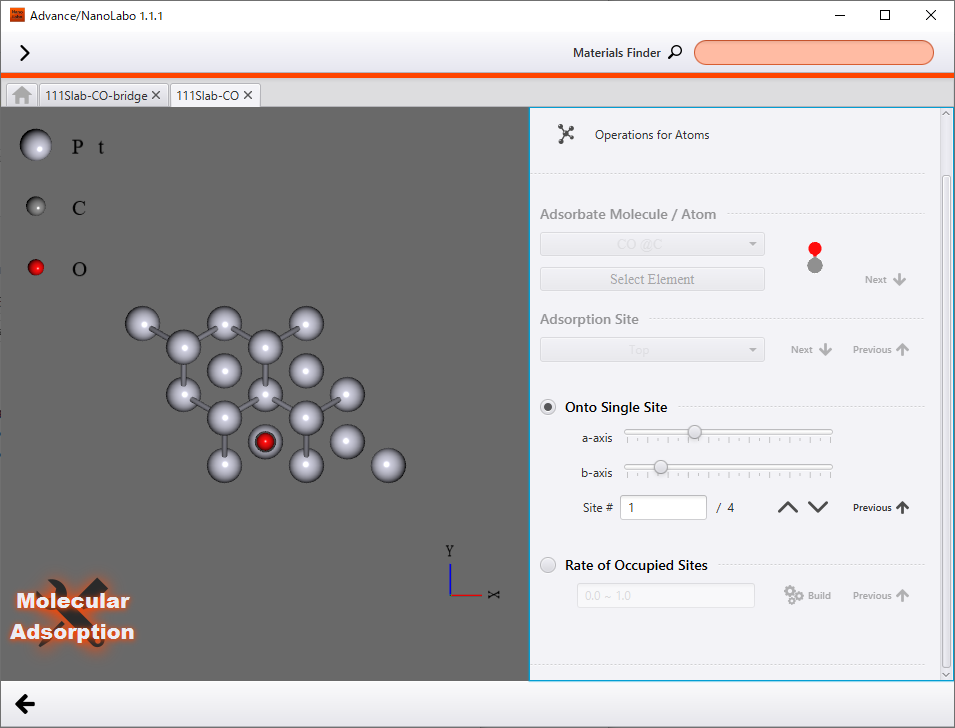
CO/Pt(111)の計算#
Pt(111)のスラブモデルのon-topサイトにCO分子を吸着させたモデル(Pt/CO(111))を作成し、構造最適化計算を行いました。また、吸着エネルギーを計算するために、孤立CO分子の構造最適化計算も行いました。Pt(111)表面にはon-topサイト以外にも複数の吸着サイトが存在しますが、低被覆率ではon-topサイトが最も安定なCO分子の吸着サイトであることが実験的に知られています4。
CO/Pt(111)・Pt(111)・CO分子の各モデルの全エネルギー2より、COの吸着エネルギーは1.45eVとなりました。吸着エネルギーの実験値は1.39eV3であり、計算値と実験値は比較的よい一致を示しています。
|

|
|
CO/Pt(111)モデルの作成 |
最適化したCO/Pt(111)の構造 |
孤立CO分子のモデル |