M3GNetを用いた界面エネルギーの計算#
M3GNetはバルク構造に最適化されていますが、今回は界面や表面について適応し、エネルギーについてDFT計算と比較しました。
バルクモデルのエネルギー計算#
表面エネルギーや界面エネルギーを計算するために、バルクのエネルギーを計算しておきます。 計算にはMaterials Project(https://materialsproject.org/)の構造を用いました。 まずAnisotropicにセルを変形させながらM3GNetで構造最適化をしたのち、 M3GNetとQuantum Espressoでそれぞれエネルギーを計算しました。
Quantum EspressoのSCF計算ではk点の数を指定できます。 今回は(セルサイズ)×(k点数)≈25となるように設定しました。
| Al | Al2O3 | AlN | Au | GaN | Pt | Si | SiC | SiO2 | Ti | TiN | TiO2 | W | ZnO |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| mp-134 | mp-1143 | mp-661 | mp-81 | mp-804 | mp-126 | mp-149 | mp-8062 | mp-8059 | mp-72 | mp-492 | mp-390 | mp-91 | mp-2133 |
スラブモデルの表面エネルギー#
解析手順#
M3GNetをいくつかの材料の表面モデルに適用しました。 上下に真空層をもつ表面構造について、セルを固定したままM3GNetで構造最適化をしたのち、 M3GNetとQuantum Espressoでそれぞれエネルギーを計算して行いました。 また、表面エネルギーはスラブ構造のエネルギーからバルクのエネルギーを原子数分差し引くことで計算しました。
構造作成#
Advance/NanoLaboの機能を使えばスラブモデルを簡単に作成することができます。 市松模様で表示されている原子は座標が固定されているので、 Advance/NanoLaboの機能を使って全ての原子の固定を解除しました。
以下の表に作成した表面構造の面方位を示しました。
| Material | Slab 1 | Slab 2 | Slab 3 |
|---|---|---|---|
| Al | (001) | (011) | (111) |
| Au | (001) | (011) | (111) |
| Pt | (001) | (011) | (111) |
| W | (001) | (011) | (111) |
| Si | (001) | (011) | (111) |
| SiC | (001) | (011) | (111) |
| SiO2 | (001) | (011) | (111) |
| TiN | (001) | (011) | (111) |
| TiO2 | (001) | (011) | (110) |
| Ti | (001) | (100) | (110) |
| AlN | (001) | (100) | (110) |
| GaN | (001) | (100) | (110) |
| ZnO | (001) | (100) | (110) |
解析結果#
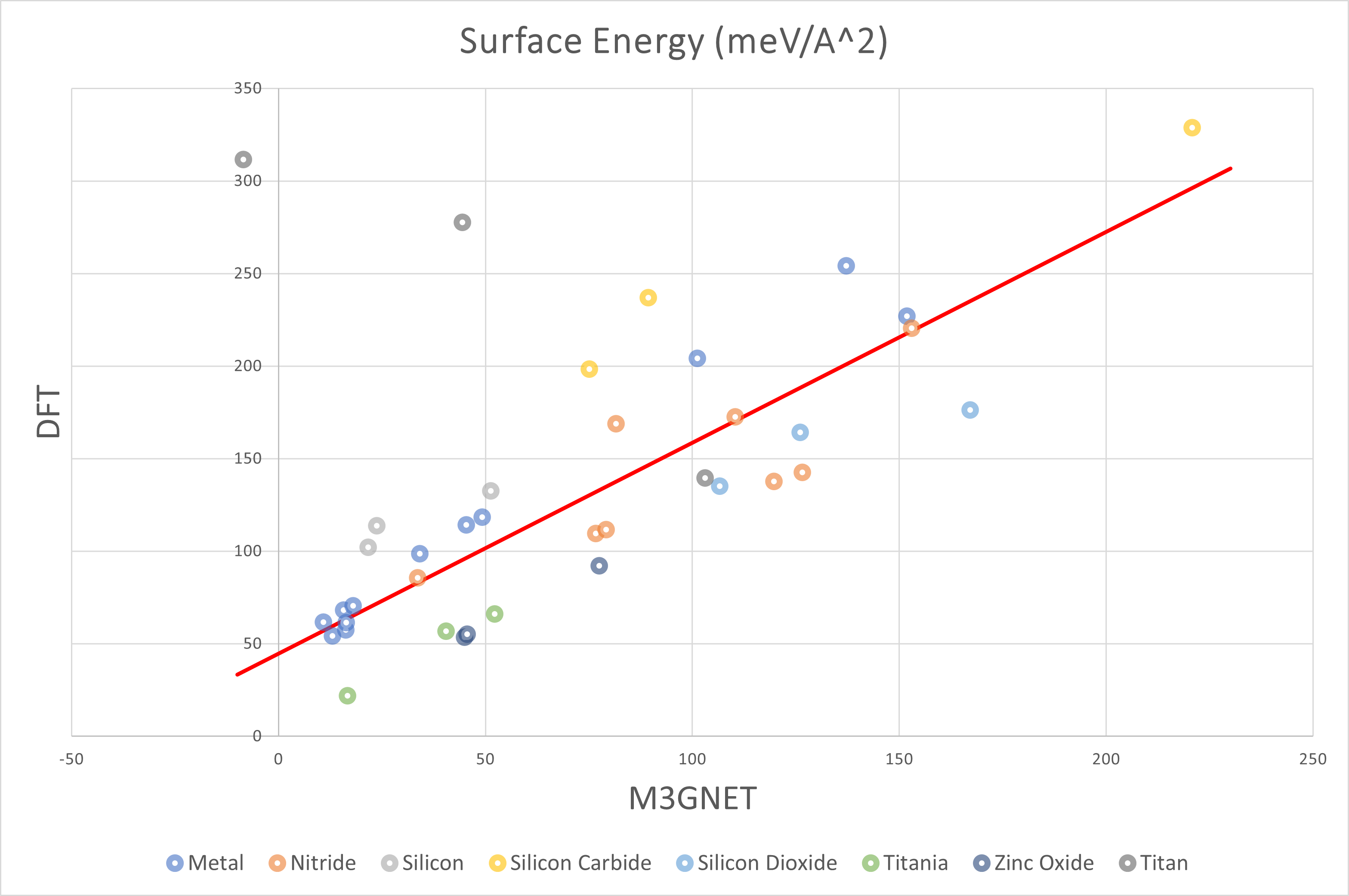
上図はDFTの結果を縦軸に、M3GNetの結果を横軸にとって表面エネルギーをプロットしたものです。 全体的な傾向として正の相関が確認できたほか、M3GNetの方がDFT計算よりも表面エネルギーを小さく見積もる傾向がみられました。
また、下の表に外れ値をとった構造や、DFT計算が収束しなかった構造をまとめました。
| Material | Slab | EM3GNet (meV/Å2) | EDFT (meV/Å2) |
|---|---|---|---|
| Ti | (001) | 44.5 | 277.5 |
| Ti | (100) | 103.2 | 139.5 |
| Ti | (110) | -8.5 | 311.6 |
| TiN | (111) | Not converged |
酸化していないTiを含むスラブモデルは外れ値を取りやすく、TiNではDFT計算が収束しませんでした。
上記の構造を除いた全構造について最小二乗法を用いた結果を以下の表に示しました。
EDFT = a EM3GNet + b (meV/Å2)
s2: 不偏分散
| Group | a | b | s |
|---|---|---|---|
| All | 1.14 | 44.70 | 36.02 |
| Metal | 1.40 | 44.71 | 13.01 |
| Nitride | 0.94 | 52.20 | 25.33 |
| Oxide | 1.10 | 8.05 | 9.43 |
界面モデルの界面エネルギー#
解析手順#
M3GNetを様々な材料の界面モデルに適用しました。 上下の真空層を取り除いた界面構造について、Anisotropicにセルを変形させながらM3GNetで構造最適化をしたのち、 M3GNetとQuantum Espressoでそれぞれエネルギーを計算して行いました。 また、界面エネルギーは界面構造のエネルギーからバルクのエネルギーを原子数分差し引くことで計算しました。
構造作成#
Advance/NanoLabo Proの機能を使えば界面モデルを簡単に作成することができます。 作成した界面モデルの真空層を薄くして、バルクとして連続となるように調整しました。
バルクモデルで示した材料を用いて、金属/金属、金属/半導体、金属/絶縁体、半導体/半導体、半導体/絶縁体、絶縁体/絶縁体それぞれについて界面モデルを作成しました。
作成した界面構造を以下の表にまとめました。
Metal
| Material 1 | Material 2 |
|---|---|
| Au(001) | Al(001) |
| Au(001) | Pt(111) |
| Ti(001) | Al(111) |
Nitride(* 異なる界面構造)
| Material 1 | Material 2 |
|---|---|
| AlN(001) | GaN(001)* |
| AlN(001) | GaN(001)* |
| AlN(001) | GaN(001)* |
| AlN(001) | SiC(111)* |
| AlN(001) | SiC(111)* |
| AlN(100) | GaN(100)* |
| AlN(100) | GaN(100)* |
| TiN(001) | W(001) |
Silicon
| material 1 | material 2 |
|---|---|
| Si(001) | SiO2(001) |
| Si(100) | Al(100) |
| Si(100) | Au(100) |
| Si(110) | Al2O3(100) |
| Si(111) | Al(111) |
| Si(111) | Au(111) |
| Si(111) | SiO2(111) |
Silicon Carbide
| material 1 | material 2 |
|---|---|
| Si(111) | SiC(111) |
Silicon Dioxide
| Material 1 | Material 2 |
|---|---|
| SiO2(100) | Al(100) |
| SiO2(100) | Au(100) |
Titania
| Material 1 | Material 2 |
|---|---|
| TiO2(001) | Au(100) |
| TiO2(001) | Ti(100) |
| TiO2(100) | Al(110) |
| TiO2(101) | Al(111) |
| TiO2(110) | Al2O3(100) |
Zinc Oxide
| Material 1 | Material 2 |
|---|---|
| ZnO(001) | Al(111) |
| ZnO(001) | Al2O3(100) |
| ZnO(110) | Al(110) |
| ZnO(110) | Pt(110) |
解析結果#
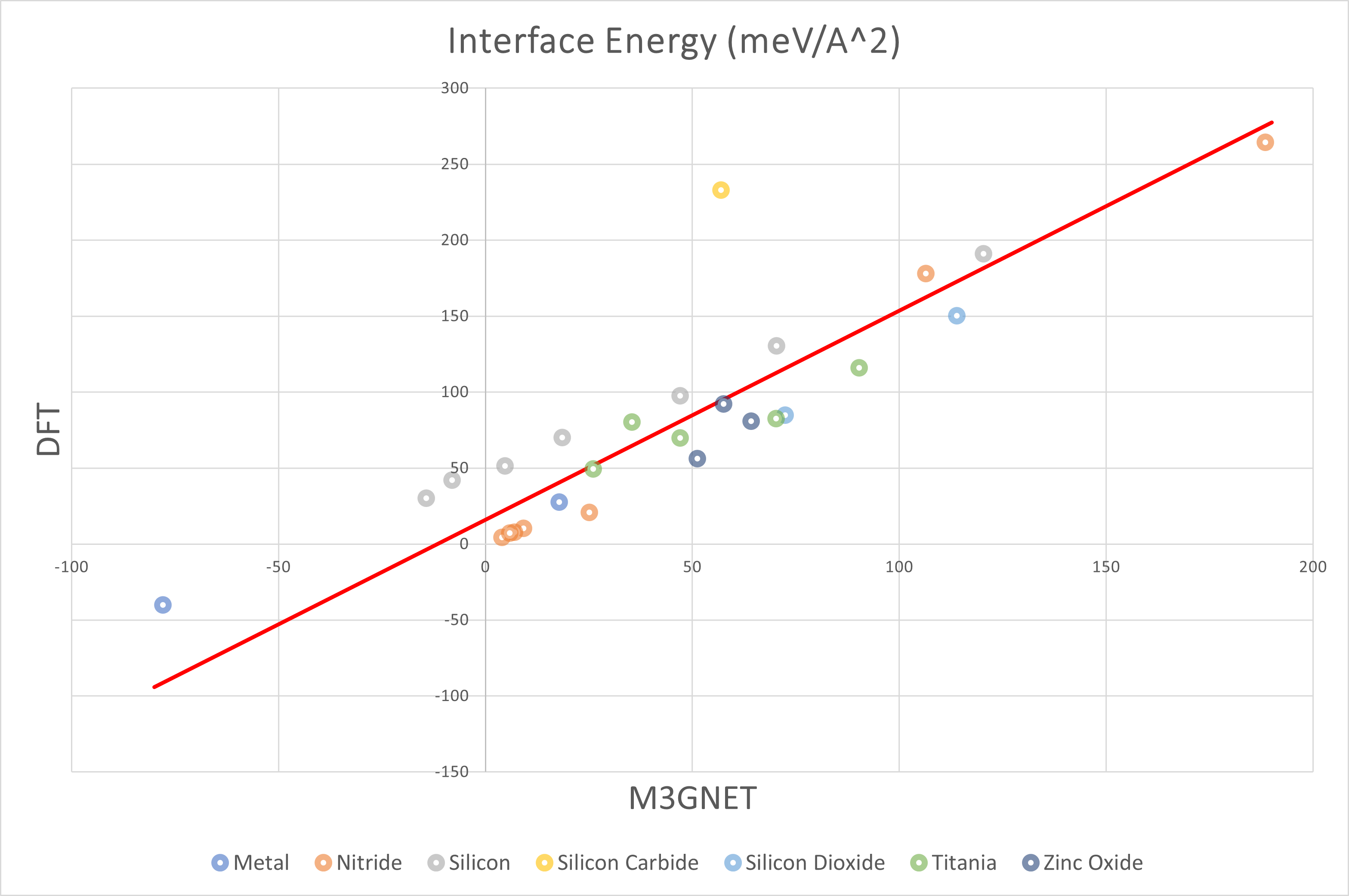
上図はDFTの結果を縦軸に、M3GNetの結果を横軸にとって界面エネルギーをプロットしたものです。 全体的な傾向として正の相関が確認できたほか、M3GNetの方がDFT計算よりも界面エネルギーを小さく見積もる傾向がみられました。
また、下の表に外れ値をとった構造や、DFT計算が収束しなかった構造をまとめました。
| Material 1 | Material 2 | EM3GNet (meV/Å2) | EDFT (meV/Å2) |
|---|---|---|---|
| Si(111) | SiC(111) | 56.9 | 232.8 |
| Ti(001) | Al(111) | -60.5 | 2185.0 |
| ZnO(110) | Pt(110) | Not converged |
表面モデルのときと同様に、酸化していないTiを含む界面モデルは外れ値を取りやすいことがわかりました。 今回はZnO(110)-Pt(110)界面モデルで計算が収束しませんでした。 また、シリコン系ではDFTとM3GNetの間に約50 meV/Å2のオフセットが生じており、 特にシリコンカーバイド系ではDFTとM3GNetの乖離が顕著に現れています。
上記の構造を除いた全構造について最小二乗法を用いた結果を以下の表に示しました。
EDFT = a EM3GNet + b (meV/Å2)
s2: 不偏分散
| Group | a | b | s |
|---|---|---|---|
| All | 1.38 | 15.85 | 25.47 |
| Silicon | 1.18 | 47.30 | 2.93 |
| Nitride | 1.55 | -4.97 | 12.47 |
| Oxide | 1.02 | 22.40 | 12.40 |
まとめ#
今回はM3GNetを用いて表面エネルギーと界面エネルギーの評価を行いました。 いずれの場合もM3GNetの方がDFTよりもエネルギーを小さく見積もる傾向がありましたが、 正の相関が観察されました。 外れ値や計算が収束しなかった構造を除いて線形回帰を行うと、 DFTとの誤差の標準偏差は表面エネルギーで約36meV/Å2、 界面エネルギーで約25meV/Å2でした。 M3GNetはバルク構造で学習させた機械学習ポテンシャルなので、 表面モデルのほうが誤差が大きくなってしまいます。
今回の結果から、M3GNetを材料候補のスクリーニングに用いることができるとわかりました。