Calculation of mechanical properties of MoS2/Ti2CT2 (T=F,O) bilayers using M3GNet#
On Advance/NanoLabo, an integrated GUI for nanomaterials, you can execute molecular dynamics simulation1 with M3GNet2, a general force field based on graph neural networks.
We introduce calculation of mechanical properties of bilayers3, promising flexible anode materials for ion batteries, via molecular dynamics simulation with M3GNet.
M3GNet, a general force field based on graph neural networks#
M3GNet is a universal interatomic potential based on graph neural networks with three-body interactions which was trained on the enormous DFT data of structural relaxation recorded in Materials Project4. M3GNet will enable you to execute molecular dynamics simulation with low computational cost, good accuracy and no need to parametrize force fields for each compound.
On Advance/NanoLabo, you can add Simple DFT-D35 correction to M3GNet to consider van der Waals force during molecular dynamics simulation. We calculated van der Waals interaction at the interface of the two layers of bilayers using Simple DFT-D3 correction.
MoS2/Ti2CT2 (T=F,O) bilayers#
Searching for flexible electrode materials is needed more and more for wearable electronics applications. Recently, it has been reported that forming heterostructures of and , which are known as two-dimensional anode materials, improve mechanical and electronic properties of those3.
We calculated mechanical properties of these materials via molecular dynamics simulation with M3GNet.
Generation of calculation models#
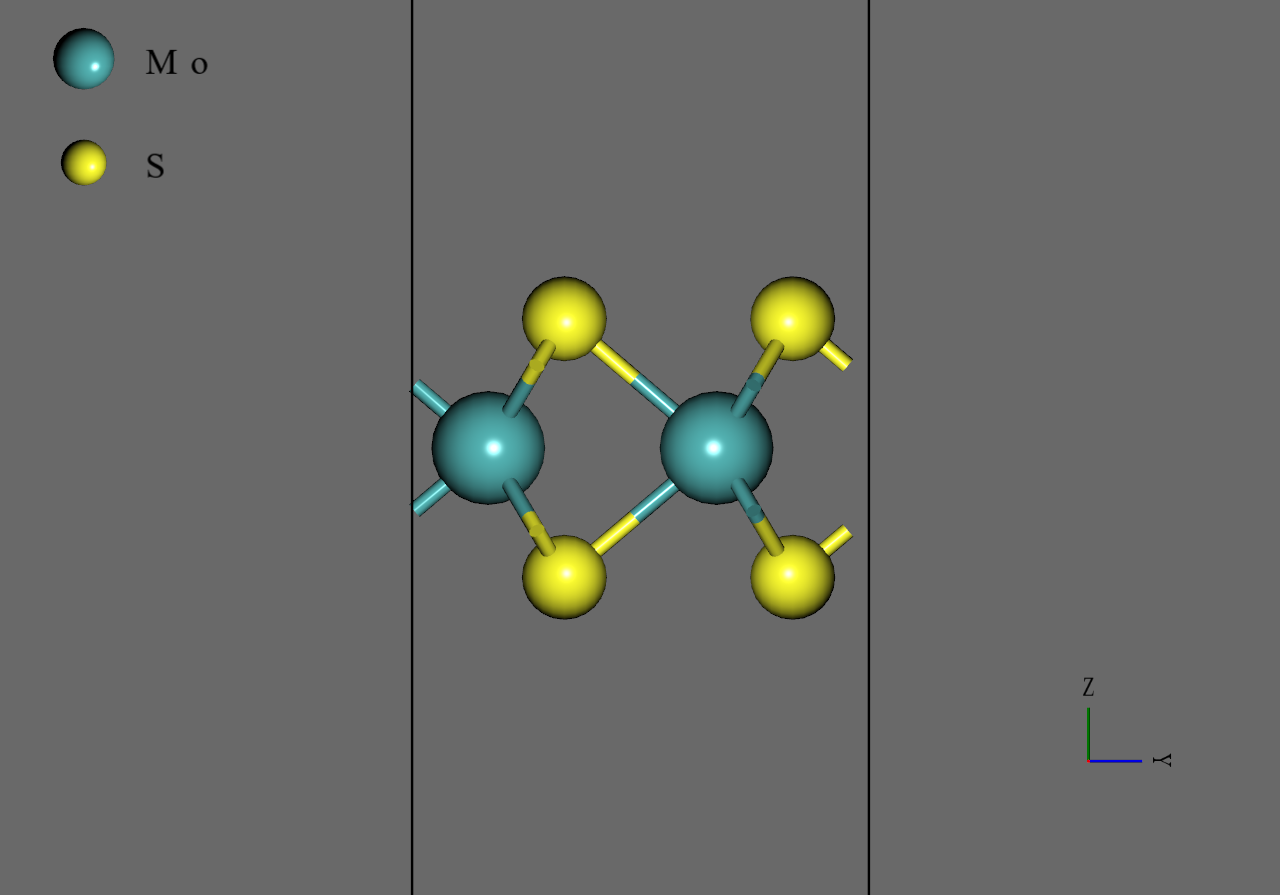
We generated model structure of from mp-2815, the structure file of downloaded from Materials Project. Firstly, we removed one layer of the bilayer of mp-2815. Secondly, we converted the cell to orthorhombic and generated supercell model. Finally, we performed structural optimization under allowing cell to change anisotropically. We note that we used M3GNet as force filed with Simple DFT-D3 correction and simulation time step was for all simulations.
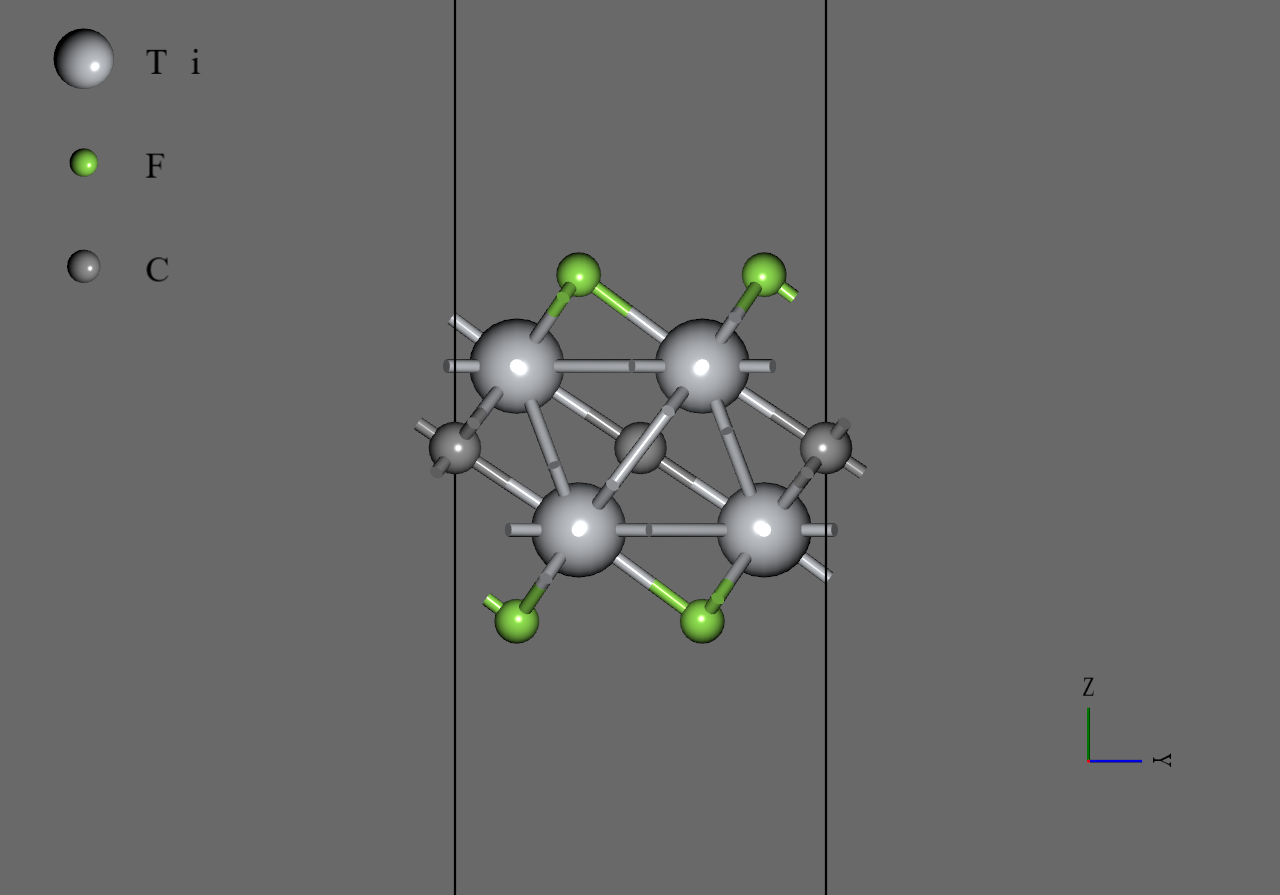
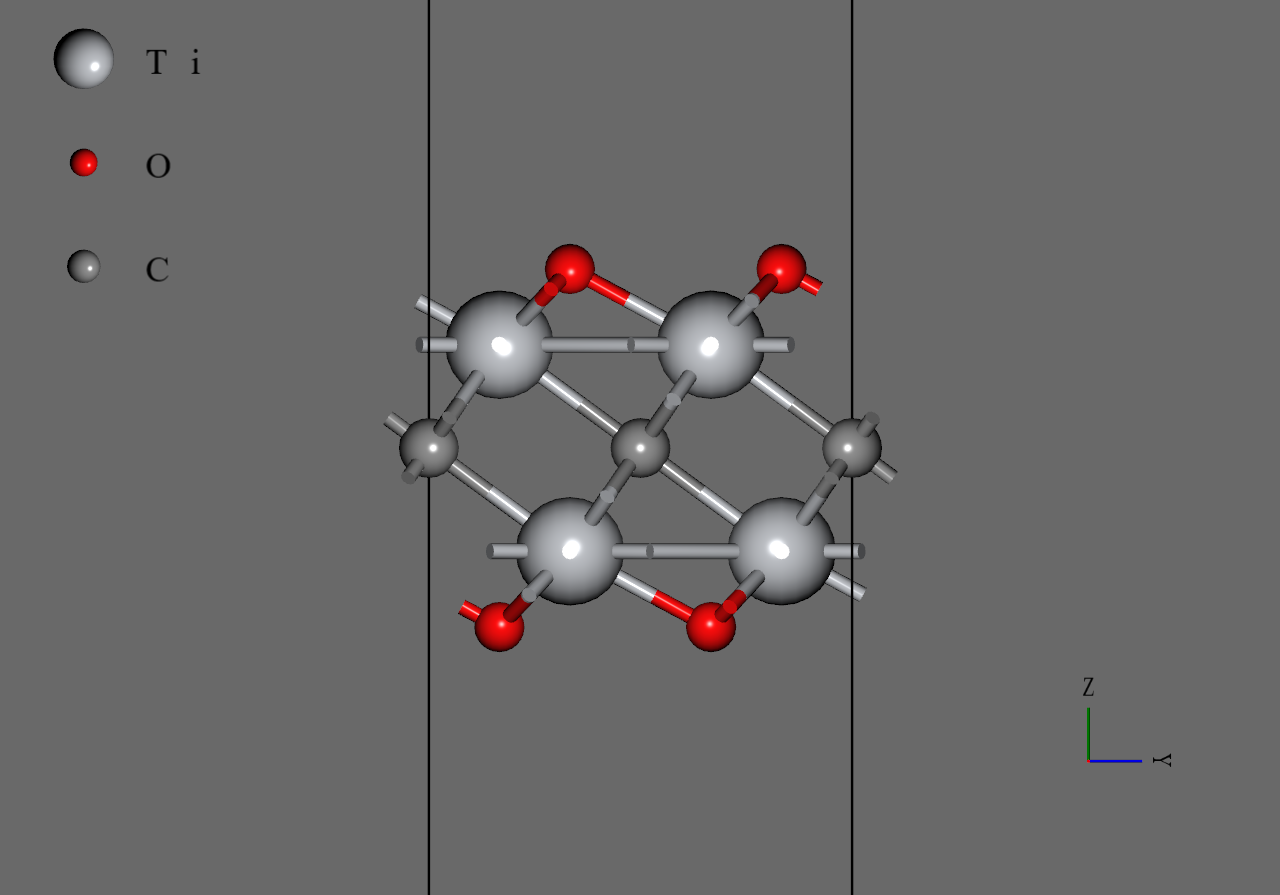
We generated model structures of from mp-12990, the structure file of 6 downloaded from Materials Project. Firstly, we removed two atoms and a atom of mp-12990 to generate monolayer of . Secondly, we replaced atoms with atoms or atoms and adjusted positions of the replaced atoms roughly. Then, we converted the cell to orthorhombic and generated supercell model. Finally, we performed structural optimization under allowing cell to change anisotropically.
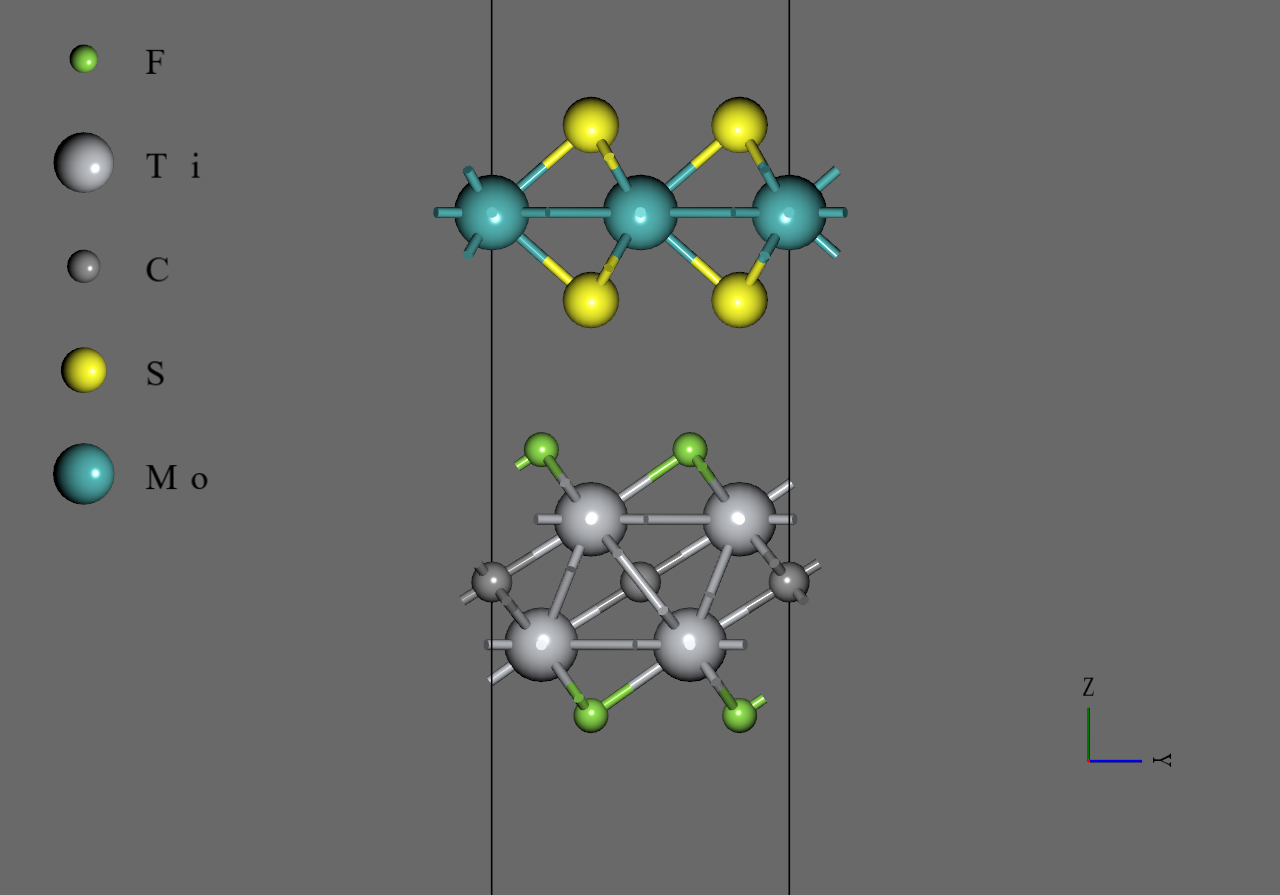
We generated model structures of bilayers from the monolayer models using interface builder function of Advance/NanoLabo. The lattice constant of the bilayer is set as the average of that of the two monolayers. In regard to the stacking patterns, atop-13 configuration was adopted for , and hcp-13 configuration was adopted for 7. Also, we adjusted interlayer distance using modeler function of Advance/NanoLabo. Finally, we performed structural optimization under allowing cell to change anisotropically.
We note that the lattice constant along direction was set as about for all models to avoid the interaction with the mirror images.
Advance/NanoLabo enables you to generate such various models on GUI easily.
|
|
|
MoS2 |
Ti2CF2 |
Ti2CO2 |
|
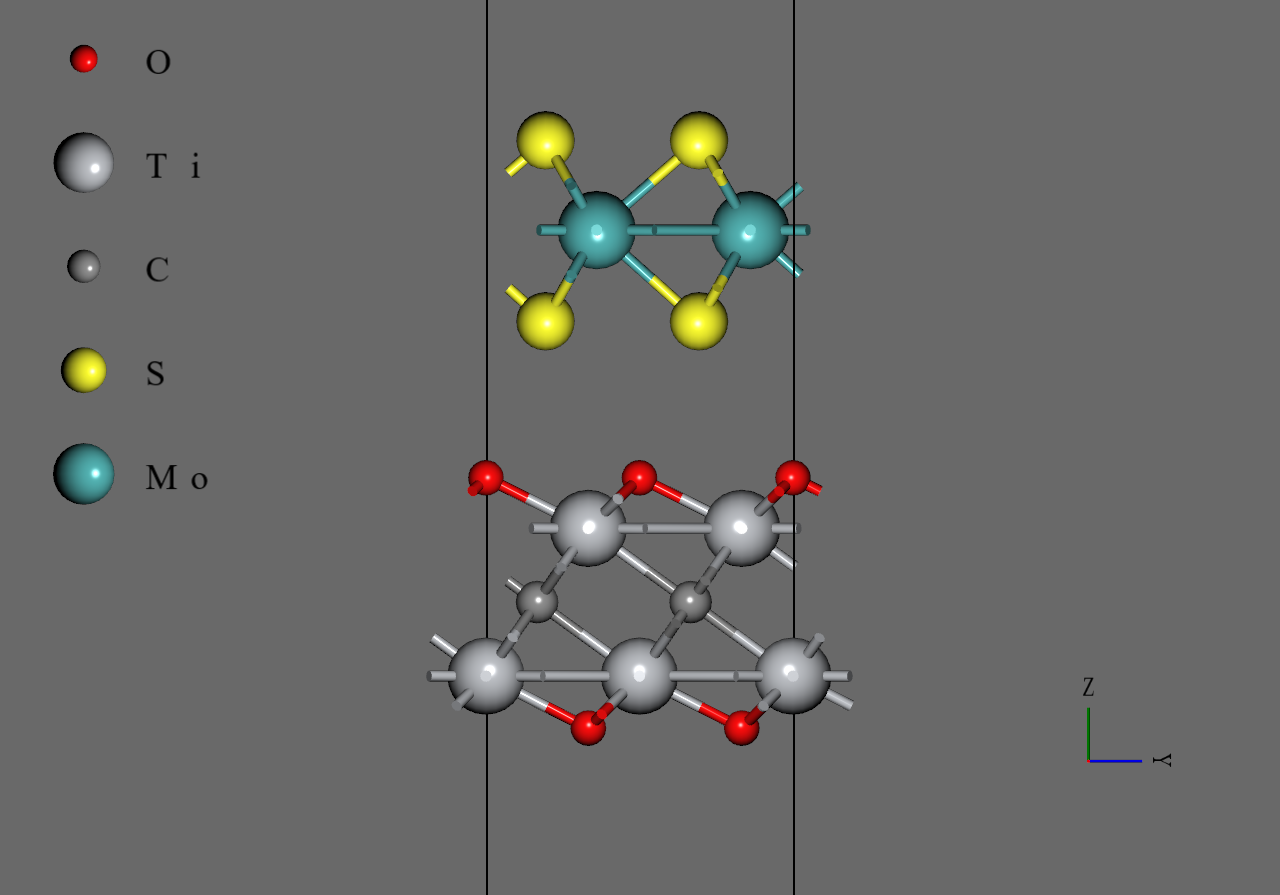
|
|
MoS2/Ti2CF2 |
MoS2/Ti2CO2 |
Setting of calculation schemes#
We alternated structural optimization and deformation along direction repeatedly for each model while calculating stress along directions. Note that directions of the all models correspond to zigzag directions of the honeycomb structures.
We executed the structural optimizations fixing the cell parameters. The strain per deformation step was set as . The structural optimization and deformation were repeated times.
You can set such a calculation scheme by editing input file directly on Advance/NanoLabo.
|
Editing of input file on GUI |
Analysis results#
We show the calculated relationships between strain and stress for each model below.
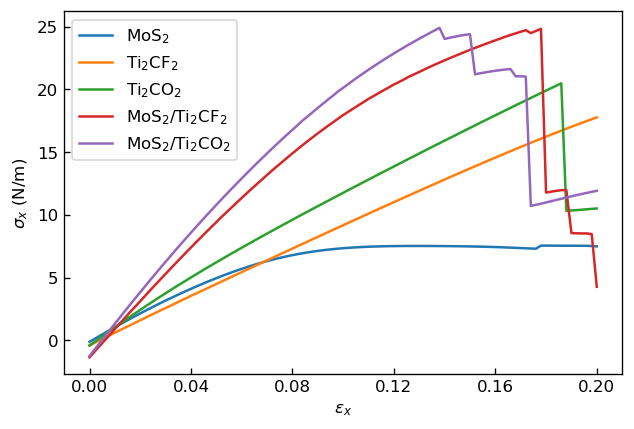
|
Relationship between strain εx and stress σx |
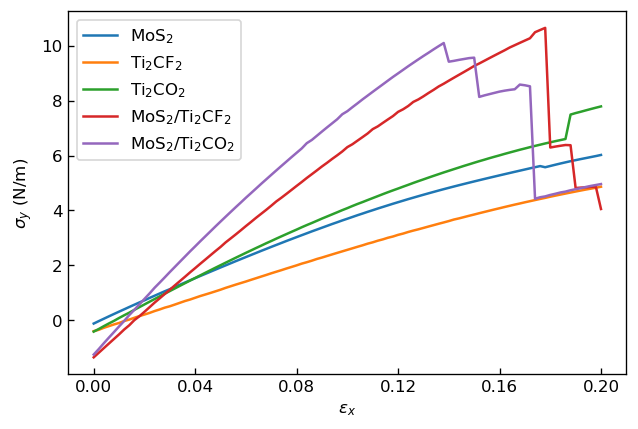
|
Relationship between strain εx and stress σy |
For two-dimensional systems without shear strain, stress and strain are related as
where are elastic constants, and due to the symmetry of the elastic constant tensor. Also, the all models are hexagonal systems, so 8.
Especially, if strain was applied only direction (), stress and are expressed as
We calculated elastic constants , by fitting linear functions to the stress-strain curves. The fitted slopes correspond to elastic constants. Note that we fitted the functions to linear regions, where strain is ~. We show the plots, in which the horizontal axes correspond to elastic constants calculated via DFT calculation in the previous research3 and the vertical axes correspond to those calculated using M3GNet.
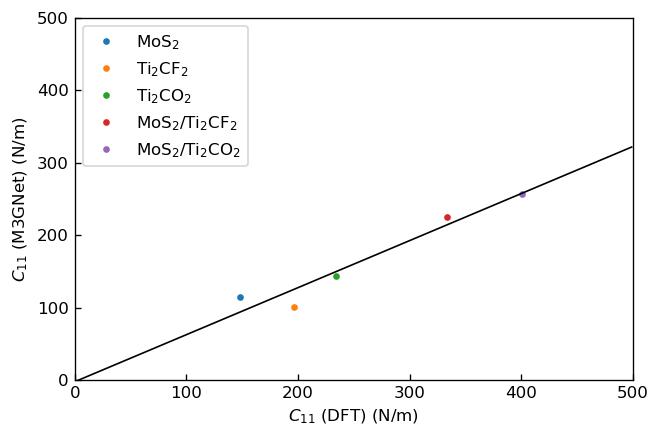
|
Correlation between C11 calculated by DFT and M3GNet |
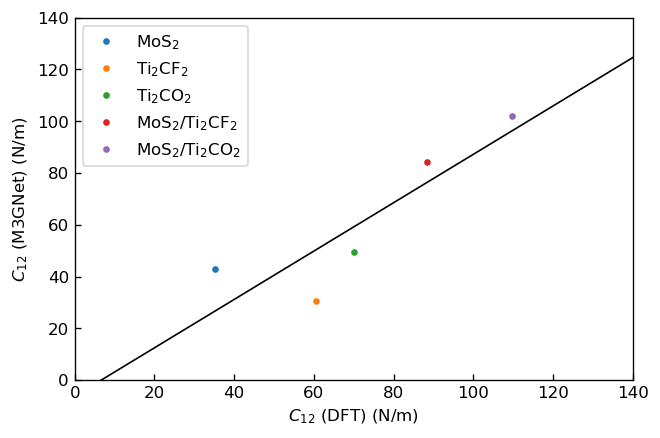
|
Correlation between C12 calculated by DFT and M3GNet |
We fitted linear functions to these plots and show the obtained fitting parameters below.
Slope |
y-intercept |
|
|---|---|---|
C11 |
0.65 |
-2 |
C12 |
0.9 |
-6 |
We can see that DFT results and M3GNet results have positive correlations for both elastic constants, but M3GNet results tend to be underestimated compared to DFT results.
We also calculated in-plane Young's modulus and Poisson's ratio of each system from the elastic constants and the following relationship3
We show the plots, in which the horizontal axes correspond to Young's modulus or Poisson's ratio calculated via DFT calculation in the previous research3 and the vertical axes correspond to those calculated using M3GNet.
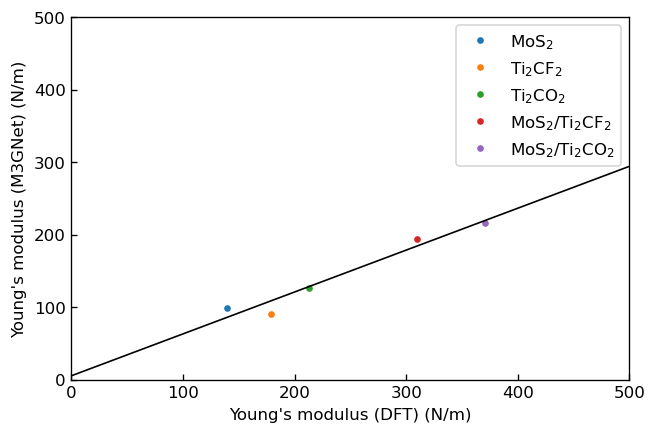
|
Correlation between Young's modulus calculated by DFT and M3GNet |
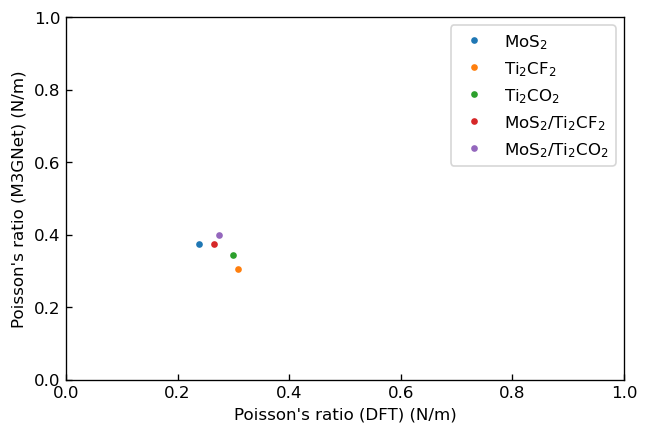
|
Correlation between Poisson's ratio calculated by DFT and M3GNet |
We fitted linear functions to these plots and show the obtained fitting parameters below.
Slope |
y-intercept |
|
|---|---|---|
E |
0.58 |
5 |
ν |
-0.9 |
0.6 |
We can see that the DFT results and the M3GNet results have positive correlations for Young's modulus , and M3GNet results tend to be underestimated compared to DFT results similarly to the elastic constants. Meanwhile, the DFT results and the M3GNet results of Poisson's ratio don't have positive correlations but have sufficient accuracy to order-estimate Poisson's ratio. Also, the M3GNet results show that formation of heterostructures improves Young's modulus for each system similarly to DFT results.
The simulation for each system was done about minutes at longest, which was executed with thread using OpenMP. It shows that simulation with M3GNet has significantly low computational cost compared to DFT calculation. Also, though M3GNet has been trained on mainly bulk structures, we obtained elastic constants of two-dimensional systems which have positive correlation with DFT results.
関連ページ#
- ナノ材料解析統合GUI Advance/NanoLabo
- 解析分野:ナノ・バイオ
- 産業分野:材料・化学
- Advance/NanoLabo Product Information
- Advance/NanoLabo Documentation
-
We used LAMMPS (Large-scale Atomic/Molecular Massively Parallel Simulator) as a solver of molecular dynamics simulation. ↩
-
Chen, C., Ong, S.P., Nat Comput Sci 2, 718–728 (2022). ↩
-
Li, Jie, et al., J. Phys. Chem. C 2019, 123, 11493−11499 ↩↩↩↩↩↩↩↩
-
https://www.materialsproject.org/ ↩
-
https://dftd3.readthedocs.io/en/latest/ ↩
-
Li, Xiao-Hong, et al. ACS Omega 2020, 5, 22248−22254 ↩
-
The atop-2 configuration of , which is adopted in the previous research3, changes to hcp-1 configuration under strain in our simulation, so we adopted hcp-1 configuration for the initial structure. ↩
-
Radevych, Danylo, Marija Gajdardziska-Josifovska, and Michael Weinert, Phys. Rev. B 107 (2023): 054104. ↩