Calculation of Density of States using ThreeBodyTB#
Overview#
ThreeBodyTB is a general-purpose tight-binding method developed by NIST1.
Generally, tight-binding methods are less accurate than first-principles calculations, but they offer the advantage of faster computational speed and applicability to relatively large systems for electronic state calculations.
However, traditional tight-binding methods required the preparation of parameters such as Hamiltonians, depending on the system, making it difficult for general-purpose applications.
In ThreeBodyTB, parameters have been pre-optimized for 65 elements, enabling general-purpose applications for a wide range of inorganic materials.
In this case study, we calculated the Density of States for Si crystals doped with impurities and GaN crystals containing point defects using ThreeBodyTB.
Additionally, we compared the computational speed of ThreeBodyTB with that of first-principles calculations.
Calculation Models and Conditions#
Calculation Models#
The calculation model consists of three cases: Si doped with Al (Si:Al), Si doped with P (Si:P), and GaN containing point defects (VGa in GaN).
For each case, we downloaded the base crystal structure from the "Materials Project" 2 and created it using Advance/Nanolabo.
The calculation models for Si:Al and Si:P were created based on mp-149 3 .
For each, we used the Element Substitution function 4 to double each axis of mp-149 and replaced one Si atom with either an Al atom or a P atom.
The calculation model for GaN containing point defects was created based on mp-804 5 .
Using the Lattice Defect function 6 , we tripled the X and Y axes of mp-804 and doubled the Z axis, then randomly removed one Ga atom.
Each calculation model underwent structural optimization using Quantum ESPRESSO after creation.
The optimized calculation models are shown below.
| Model Names | Si:Al | Si:P | VGa in GaN |
|---|---|---|---|
| Calculation Models |  |
 |
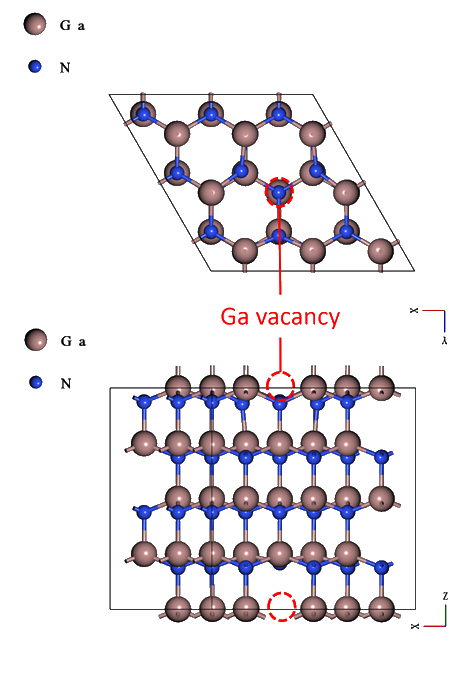 |
| Structures | Diamond structured | Diamond structured | Wurtzite structured |
| Number of Atoms | 64 (Si: 63, Al: 1) | 64 (Si: 63, P: 1) | 71 (Ga: 35, N: 36) |
Calculation Conditions#
Calculation Settings#
| Items | ThreeBodyTB | Quantum ESPRESSO |
|---|---|---|
| Wavefunction Cutoff Energy (Ry) | - | 40.0 |
| Electron Density Cutoff Energy (Ry) | - | 360.0 |
| SCF Calculation Convergence Threshold (Ry) | 1.0e-6 | 1.0e-6 |
| k-points (SCF) | 4 x 4 x 4 | 4 x 4 x 4 |
| k-points (DOS) | 8 x 8 x 8 | 8 x 8 x 8 |
Calculation Environment#
| Items | Details |
|---|---|
| CPU | Intel Xeon Gold 6330 (28 Core, 2.00 GHz) |
| Memory | 128 GB |
| Parallelism | 16 |
Density of States in Si Crystals with Different Dopants#
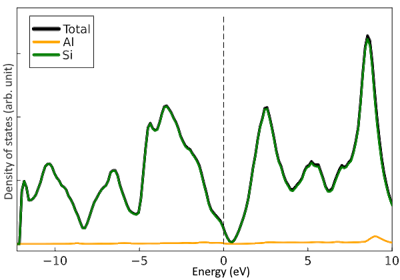
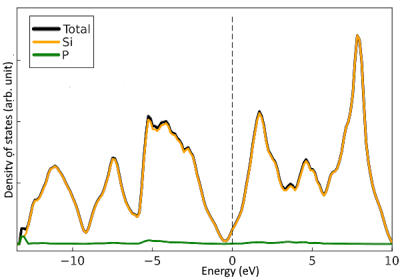
The results of calculating the density of states for Si:Al and Si:P using ThreeBodyTB are presented.
Si:Al is p-type, causing the Fermi level to shift towards higher energy, while Si:P is n-type, causing the Fermi level to shift towards lower energy.
In the calculation results as well, the Fermi level for Si:Al shifted towards higher energy, and the Fermi level for Si:P shifted towards lower energy.
| Si:Al | Si:P |
|---|---|
 |
 |
Comparison of Calculation Results between ThreeBodyTB and Quantum ESPRESSO#
Comparison of Density of States#
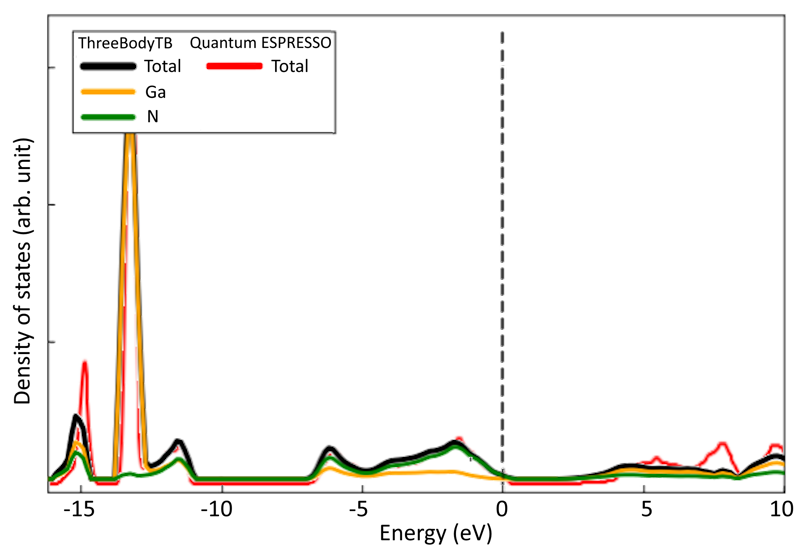
The density of states for VGa in GaN is presented.
At energies lower than the Fermi level, the peaks from ThreeBodyTB and Quantum ESPRESSO nearly coincided.
On the other hand, at energies 7-8 eV higher than the Fermi level, the broad peak appearing in Quantum ESPRESSO was not accurately calculated by ThreeBodyTB.
Comparison of Calculation Times#
The calculation time for Quantum ESPRESSO and ThreeBodyTB with 16 parallel processes for each model is presented.
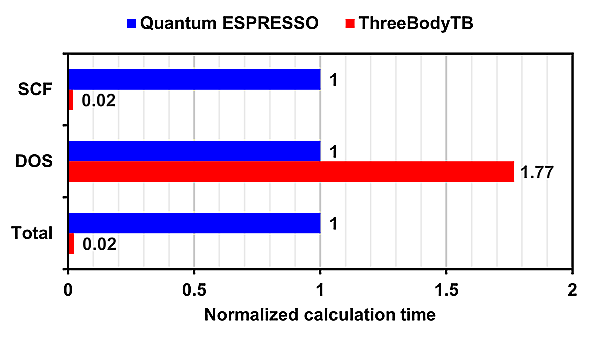
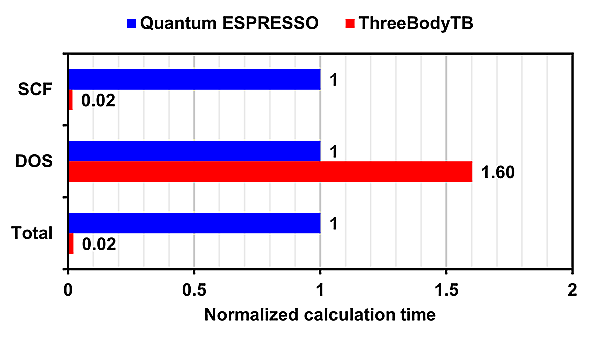
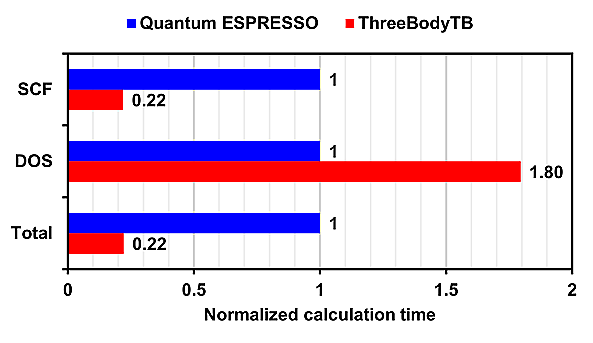
Additionally, the normalized calculation time, with Quantum ESPRESSO's time set as 1, is also shown.
For SCF (Self-Consistent Field) calculations, ThreeBodyTB was overwhelmingly faster, while for DOS (Density of States) calculations, Quantum ESPRESSO was faster.
Since DOS calculations finish in a shorter time than SCF calculations, the overall calculation time is dominated by the time taken for SCF calculations.
In calculations for Si:Al and Si:P, where all atomic sites are filled, the calculation time for ThreeBodyTB was approximately 1/50 of that for Quantum ESPRESSO.
On the other hand, in calculations for GaN containing a single vacancy, the calculation time for ThreeBodyTB was approximately 1/5 of that for Quantum ESPRESSO.
In all calculation models, ThreeBodyTB had a shorter calculation time than Quantum ESPRESSO.
| Actual Calculation Times | Normalized Calculation Times (Quantum ESPRESSO = 1) | |
|---|---|---|
| Si:Al | 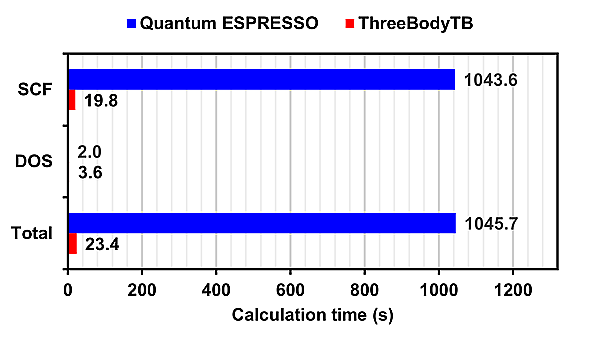 |
 |
| Si:P | 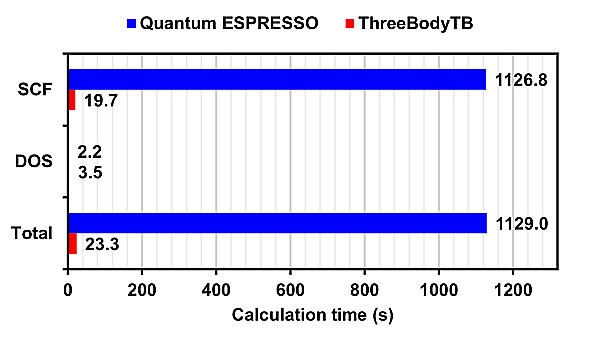 |
 |
| VGa in GaN | 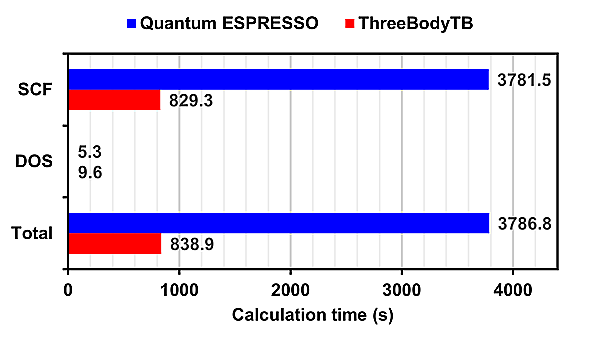 |
 |
Conclusion#
We calculated the density of states for Si crystals doped with Al or P and GaN crystals containing point defects at Ga sites using ThreeBodyTB.
In the density of states calculations for Si doped with Al or P, we confirmed that the Fermi level shifts due to impurity doping could be calculated.
In the GaN calculations, we compared the results of density of states calculations between ThreeBodyTB and Quantum ESPRESSO.
While the density of states was not accurately calculated at energies 7-8 eV higher than the Fermi level, the peak positions nearly coincided at energies lower than the Fermi level.
Comparing the calculation speeds between ThreeBodyTB and Quantum ESPRESSO, we found that SCF calculations were overwhelmingly faster with ThreeBodyTB, while DOS calculations were faster with Quantum ESPRESSO.
Overall, the calculation time was dominated by the time for SCF calculations, and we confirmed that ThreeBodyTB was faster than Quantum ESPRESSO in all models.
関連ページ#
- ナノ材料解析統合GUI Advance/NanoLabo
- 解析分野:ナノ・バイオ
- 産業分野:材料・化学
- Advance/NanoLabo Product Information
- Advance/NanoLabo Documentation
-
NIST, Home · ThreeBodyTB.jl Documentation. Available: https://pages.nist.gov/ThreeBodyTB.jl/ ↩
-
The Materials Project, Materials Project - Home. Available: https://next-gen.materialsproject.org/ ↩
-
The Materials Project, mp-149: Si (Cubic, Fd-3m, 227). Available: https://next-gen.materialsproject.org/materials/mp-149 ↩
-
AdvanceSoft, Modeler — Advance/NanoLabo documentation. Available: https://nanolabo-doc.readthedocs.io/en/latest/usage/modeler.html#substitution. ↩
-
The Materials Project, mp-804: GaN (Hexagonal, P6_3mc, 186). Available: https://next-gen.materialsproject.org/materials/mp-804 ↩
-
AdvanceSoft, Modeler — Advance/NanoLabo documentation. Available: https://nanolabo-doc.readthedocs.io/en/latest/usage/modeler.html#defect ↩