汎用グラフニューラルネットワーク力場M3GNetを用いたLi3OCl1-xBrxの室温におけるイオン伝導率の計算#
ナノ材料統合GUI Advance/NanoLaboでは、汎用グラフニューラルネットワーク力場M3GNet1を用いた分子動力学シミュレーション2を実行することができます。 本事例では、M3GNetを用いた分子動力学シミュレーションを行い、リチウムイオン伝導体の室温におけるイオン伝導率の組成依存性を計算しました。
汎用グラフニューラルネットワーク力場M3GNet#
M3GNetは、Materials Project3の膨大な構造緩和データを学習した、3体相互作用を伴う汎用グラフニューラルネットワーク力場です。 Materials Projectにおける構造緩和はDFT計算によって行われています。学習には62,783種類の化合物のデータを用いており、エネルギー、力、応力のそれぞれについて187,687件、16,875,138件、1,689,183件のデータセット(MPF.2021.2.8)を使用しています。
M3GNetを用いることで、物質ごとに力場パラメータの最適化をすることなく、分子動力学計算を短時間で精度よく実行できることが期待されます。
リチウムイオン伝導体Li3OCl1-xBrx#
は固体のリチウムイオン伝導体であり、第一原理分子動力学によるとの組成4で最も高いイオン伝導率((室温))を示すことが報告されています5。本事例では、M3GNetを用いた分子動力学計算により、のイオン伝導率が組成によってどのように変化するのかを計算しました。
計算モデルの作成#
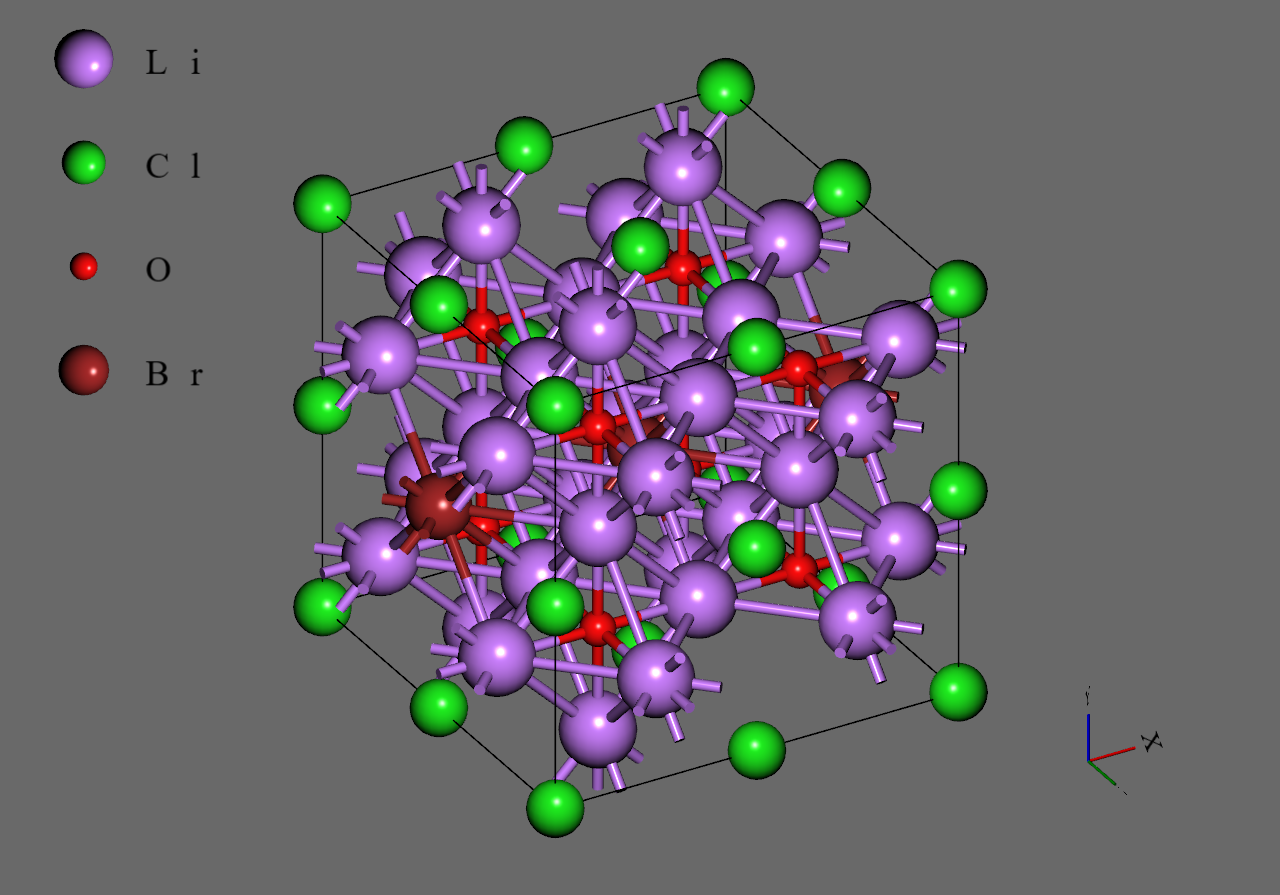
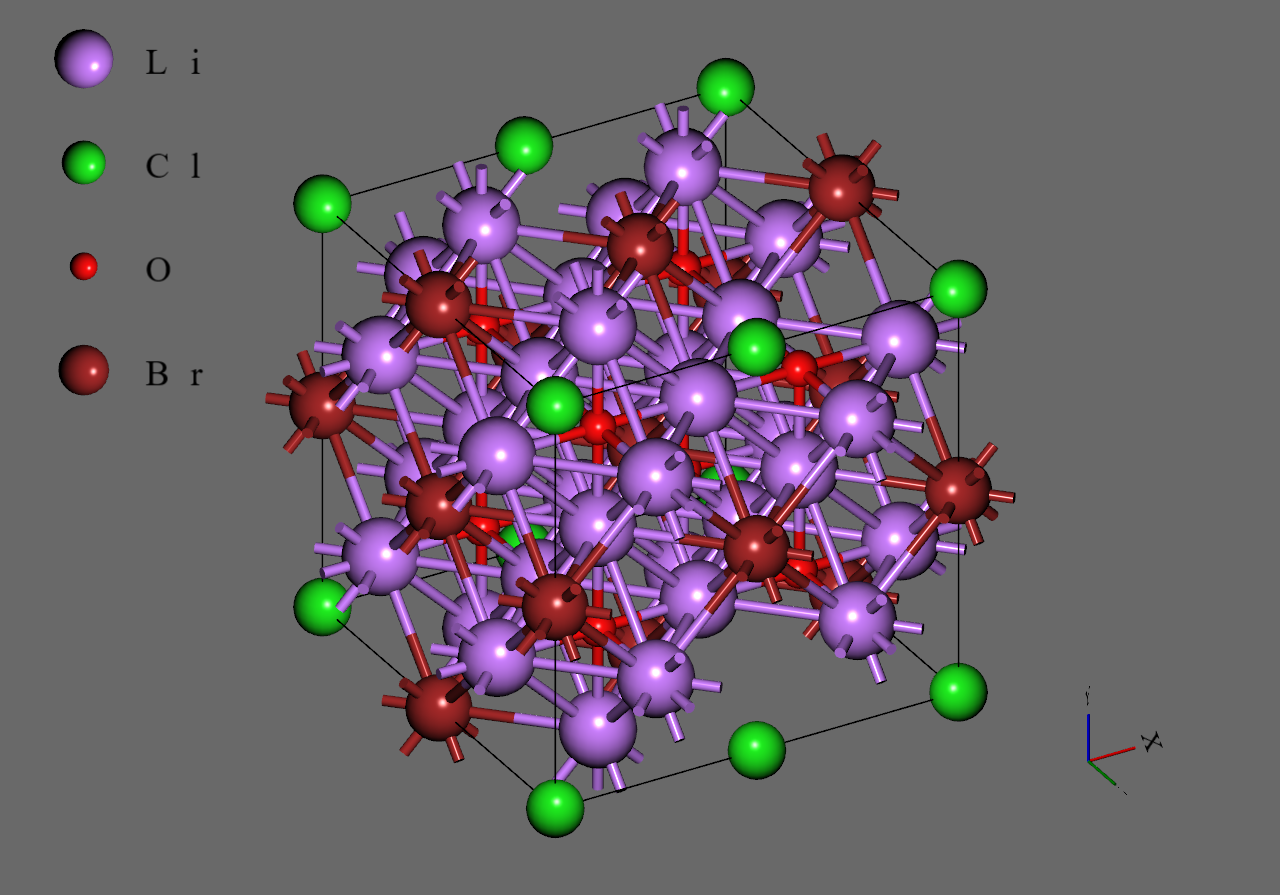
まず初めに、Materials Projectより取得した のユニットセル(mp-985585)のスーパーセルモデルを作成しました。次に、をに置換し、のそれぞれに対応するモデルを作成しました。のモデルでは、それぞれにおいて系の対称性が最も高くなるように元素の置換を行いました6。最後に各モデルから1つのを取り除き、空孔を導入しました5。
Advance/NanoLaboでは、GUIによりこれらのようなモデルを容易に作成することができます。

|
|

|
Li3OCl |
Li3OCl0.75Br0.25 |
Li3OCl0.5Br0.5 |
|

|
|
Li3OCl0.25Br0.75 |
Li3OBr |
計算スキームの設定#
以降のすべてのシミュレーションにおいて、力場にはM3GNetを用いて計算を行いました。
まず、各モデルについて、の条件下でセルに異方的な変形を許して構造最適化を行いました。このとき、時間の刻み幅はで計算を行いました。
次に、の割合で一定の温度(各組成ごとに)まで系の温度を昇温し、その後ステップ()の一定温度のシミュレーションを行いました。
最後に、各温度のモデルについて、MSD(Mean Square Displacement)の計算をステップ()行いました。
これらの動力学計算では、統計的アンサンブルにNVTアンサンブルを採用しています。また、シミュレーション時間の刻み幅はとしました。
Advance/NanoLaboではDiffusion CoefficientスイッチをONにするだけで各元素のMSDの計算・可視化を行うことができます。
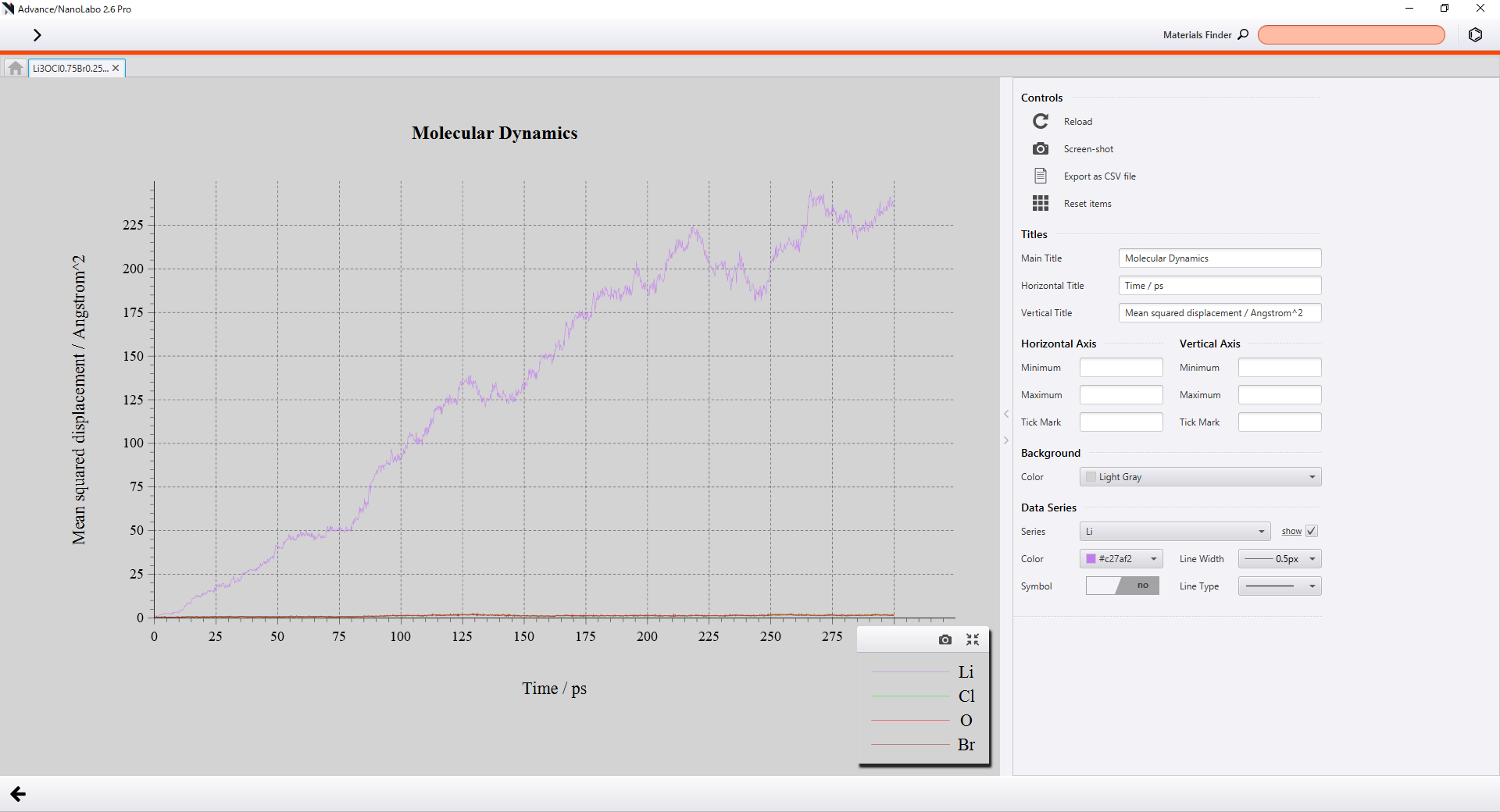
|
GUI上でのMSDの可視化 |
計算結果#
3次元系の場合、拡散係数とMSDは次の関係にあります。
ここでは時間を表します。 この関係式を用いてMSDの時間変化のプロットをフィッティングし、その傾きからの拡散係数を算出しました。各組成・各温度において求められた拡散係数のアレニウスプロットを下図に示します。
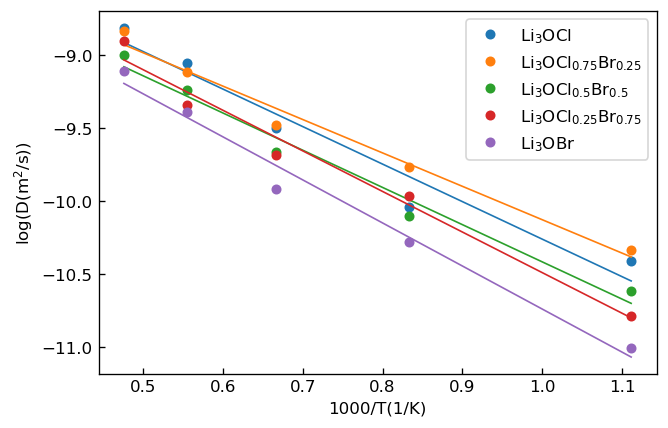
|
拡散係数Dのアレニウスプロット |
また、イオン伝導率は、拡散係数と温度を用いて次のように表されます。
ここで、はイオンの数密度、は素電荷、はボルツマン定数、は活性化エネルギーを表します。 まず初めに、この関係式を用いてアレニウスプロットをフィッティングし、各組成における活性化エネルギーおよび定数を算出しました。 次に、算出したおよびを用いて、における各組成のイオン伝導率を計算しました。その結果を下図に示します。 図からは、先行研究の第一原理分子動力学の結果5と同様にして、をから増加させるとのときにイオン伝導率は最大となり、その後は単調に減少してで最小となることがわかります。ただし、イオン伝導率の絶対値は第一原理分子動力学の結果に比べて小さく見積られています。この原因としては、高温においてはM3GNetの計算精度が下がり、拡散係数を過大評価している可能性などが考えられます。

|
Li3OCl1-xBrxのイオン伝導率の組成依存性 |
本事例において、一つの組成・一つの温度についてのシミュレーションにかかった時間は並列化をせずに約2時間程度であり、第一原理分子動力学に比べて大幅に計算コストが削減されています。 それにも関わらず、第一原理分子動力学で得られたイオン伝導率の組成依存性と同様の傾向を示す計算結果を得ることができました。
関連ページ#
-
Chen, C., Ong, S.P. A universal graph deep learning interatomic potential for the periodic table. Nat Comput Sci 2, 718–728 (2022). ↩
-
ソルバーにはLAMMPS(Large-scale Atomic/Molecular Massively Parallel Simulator)を用いています。 ↩
-
Materials Projectは物質材料のオンラインデータベースです。カリフォルニア大学バークレー校のKristin Persson准教授らのグループによって運用されています ↩
-
1つの空孔を導入した2×2×2スーパーセルモデル(Li23O8Cl8(1-x)Br8x)における計算結果 ↩
-
Deng, Z., Radhakrishnan, B., and Ong, S. P., Chemistry of Materials 2015 27 (10), 3749-3755 ↩↩↩
-
Chen, Ronghan, et al., ACS Applied Energy Materials 2021 4 (3), 2107-2114 ↩