Calculation of ionic conductivity at 300 K of Li3OCl1-xBrx with M3GNet, a general force field based on graph neural networks#
On Advance/NanoLabo, an integrated GUI for nanomaterials, you can execute molecular dynamics simulation1 with M3GNet2, a general force field based on graph neural networks.
We introduce calculation of composition dependence of ionic conductivity at 300 K of a lithium superionic conductor via molecular dynamics simulation with M3GNet.
M3GNet, a general force field based on graph neural networks#
M3GNet is a universal interatomic potential based on graph neural networks with three-body interactions which was trained on the enormous data of structural relaxation recorded in Materials Project3.
Structural relaxation in Materials Project was executed via DFT calculation. M3GNet was trained on MPF.2021.2.8, a dataset which contains 62,783 compounds with 187,687 energies, 16,875,138 force components and 1,689,183 stress components.
M3GNet will enable you to execute molecular dynamics simulation with low computational cost, good accuracy and no need to parametrize force fields for each compound.
Li3OCl1-xBrx, a lithium superionic conductor#
is a solid lithium superionic conductor. It is reported5 that ab initio molecular dynamics found 4 is the highest conductivity composition.
We calculated composition dependence of ionic conductivity of via molecular dynamics simulation with M3GNet.
Generation of calculation models#
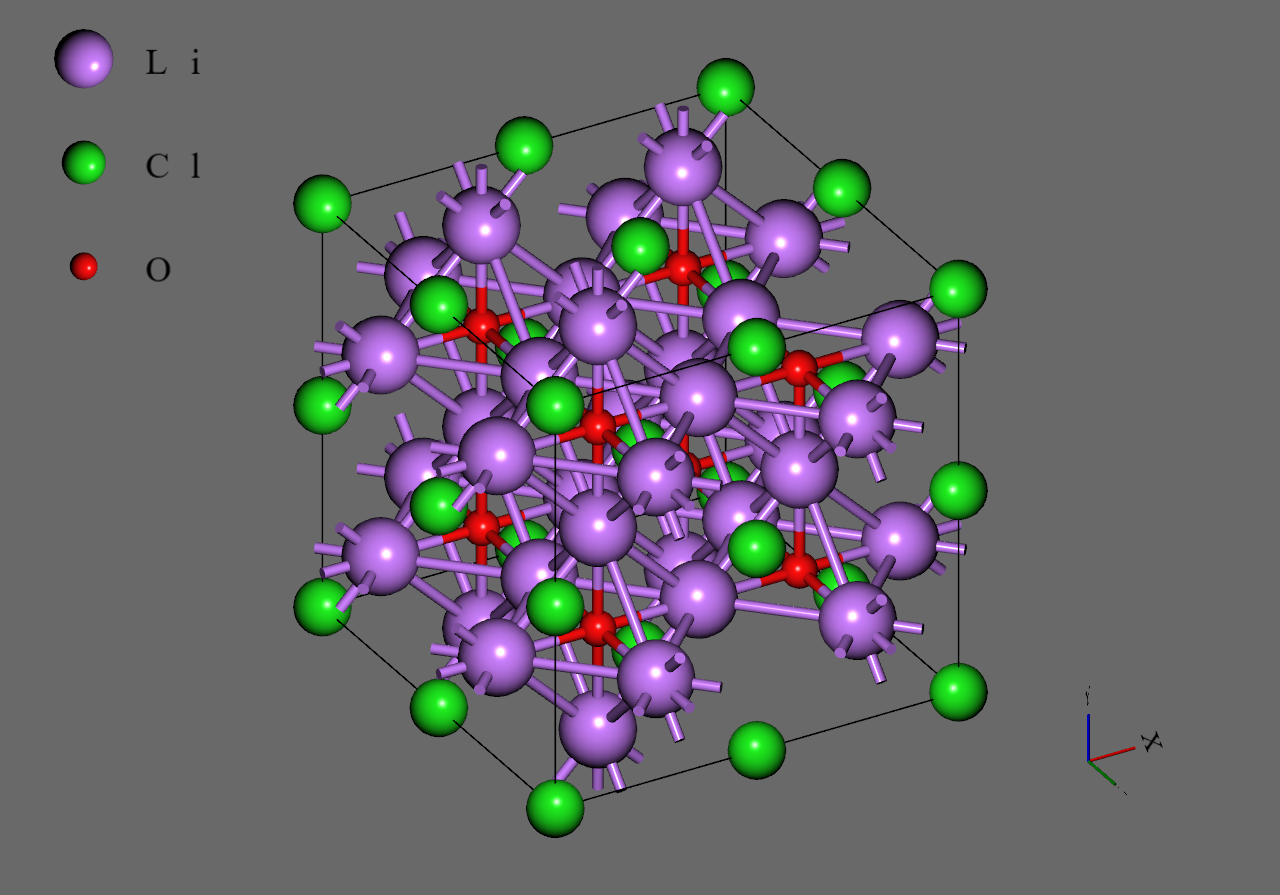
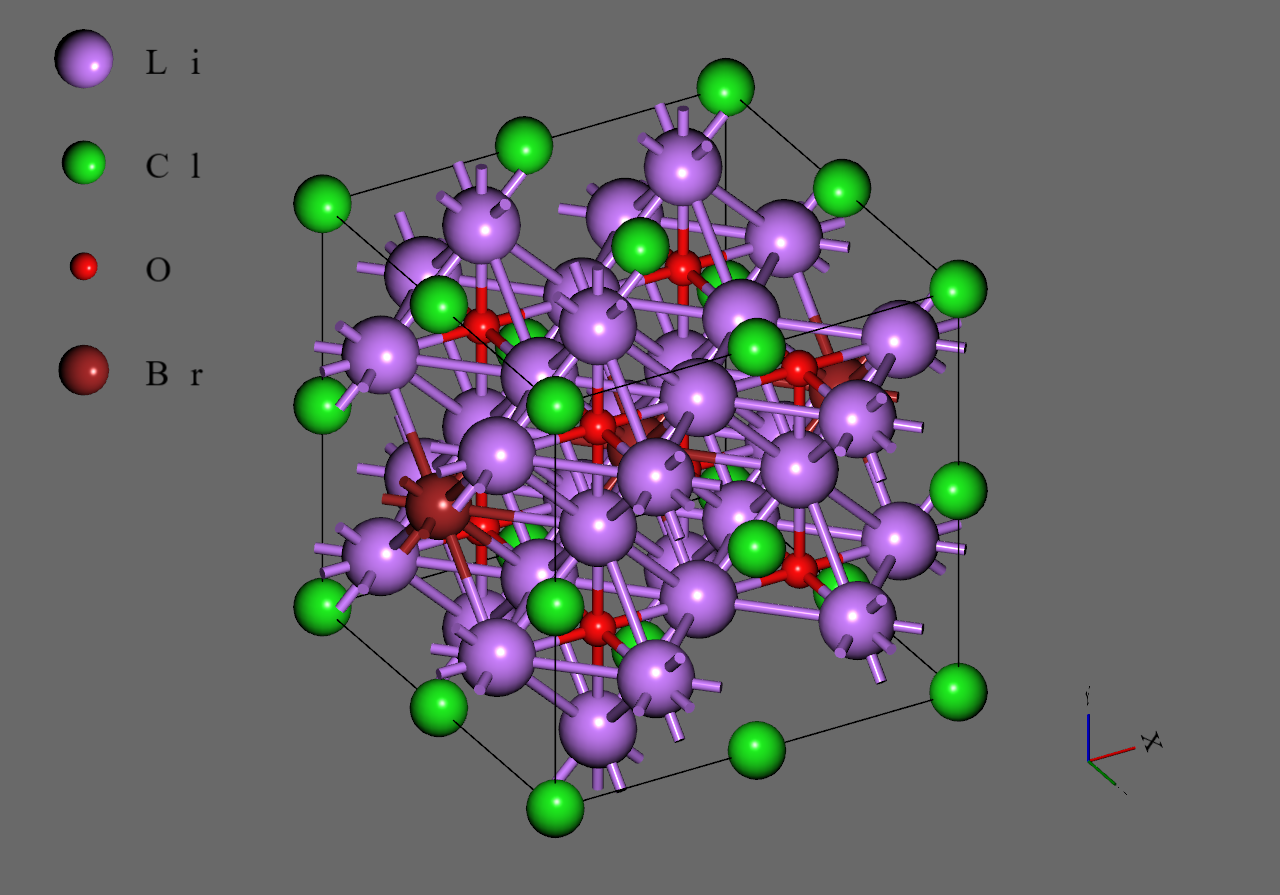
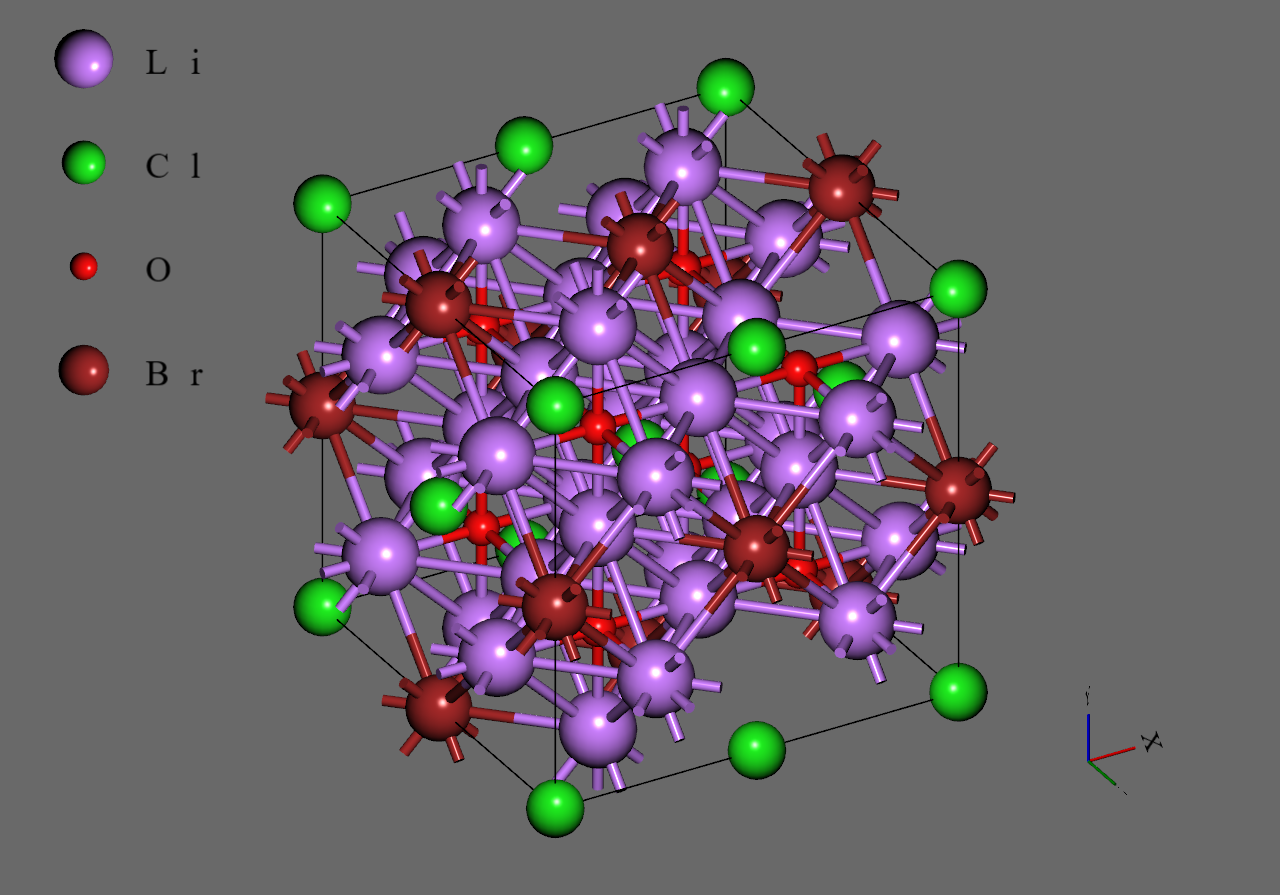
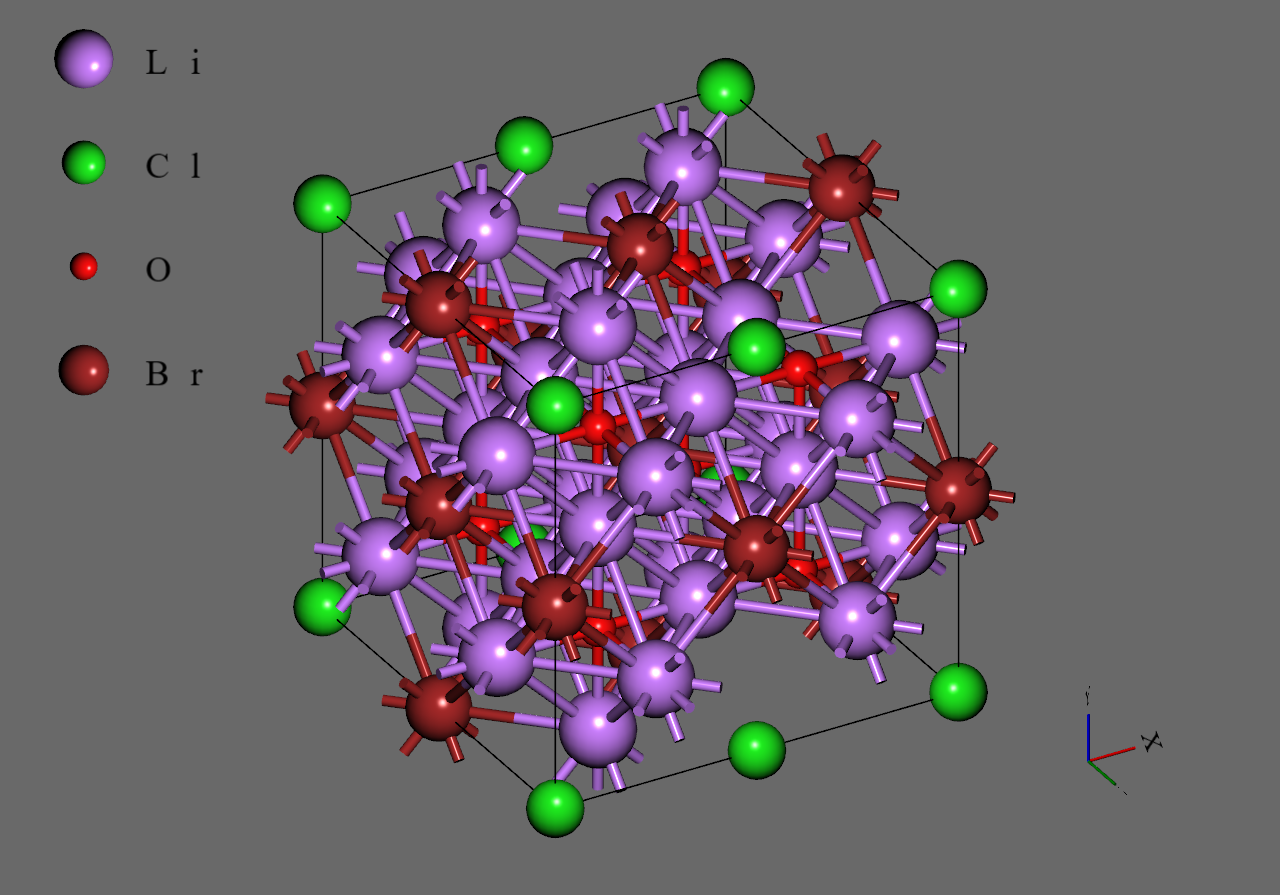
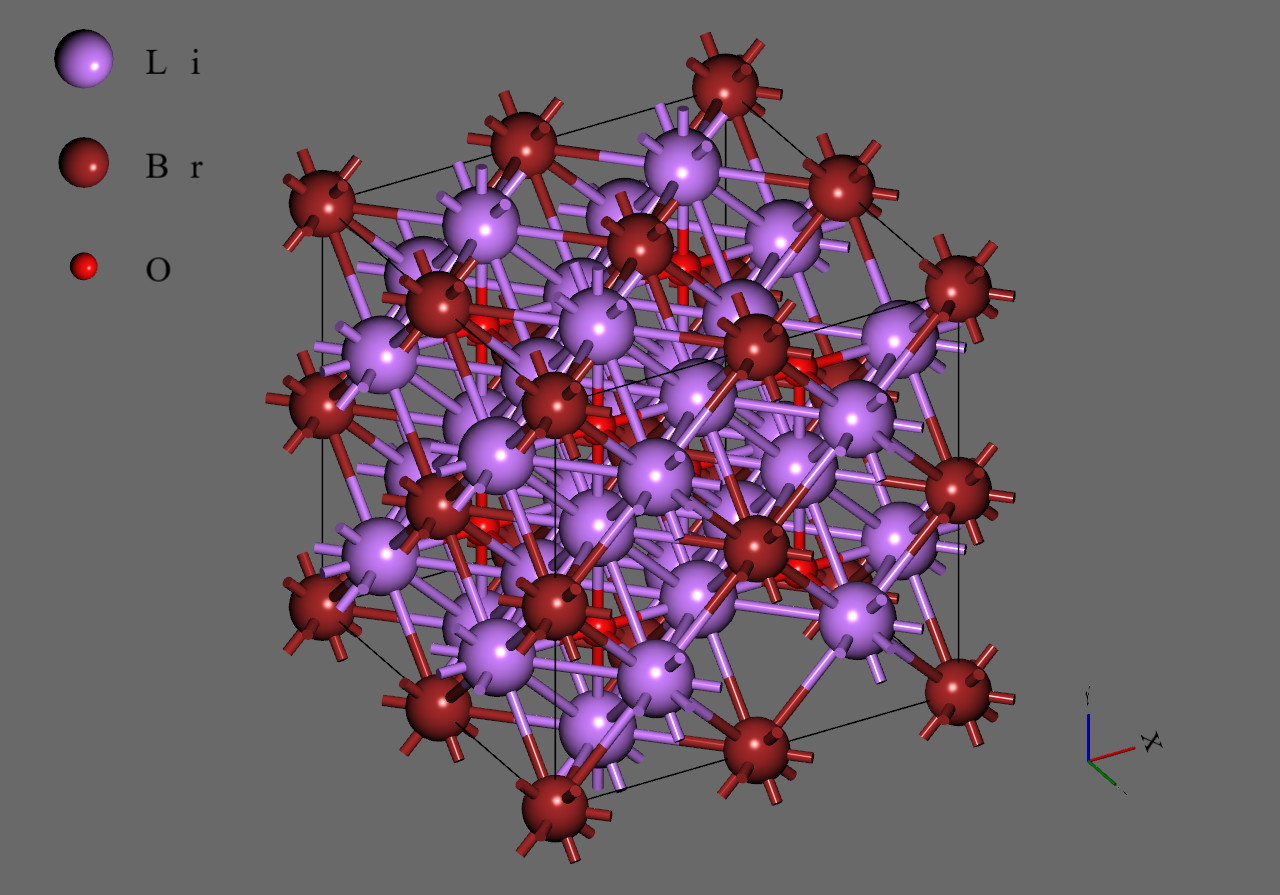
Firstly, we generated supercell models from the unit cell model of (mp-985585) downloaded from Materials Project
Secondly, we replaced with to construct models corresponding to and , respectively. We adopted structures with highest symmetry for and , respectively6.
Finally, we introduced single vacancy for each model by removing a atom5.
Advance/NanoLabo enables you to generate such models on GUI easily.
|
|
|
Li3OCl |
Li3OCl0.75Br0.25 |
Li3OCl0.5Br0.5 |
|
|
|
Li3OCl0.25Br0.75 |
Li3OBr |
Setting of calculation schemes#
We used M3GNet as force filed for all simulations.
First, we performed structural optimization under allowing cell to change anisotropically for each model. Simulation time step was .
Next, we heated each system up to the desired temperature at rate of and preformed isothermal molecular dynamics simulation for steps ().
And finally, we calculated MSD (Mean Square Displacement) of systems at each temperature for steps ().
We employed NVT ensemble as statistical ensemble for these dynamics calculation. We set simulation time step as .
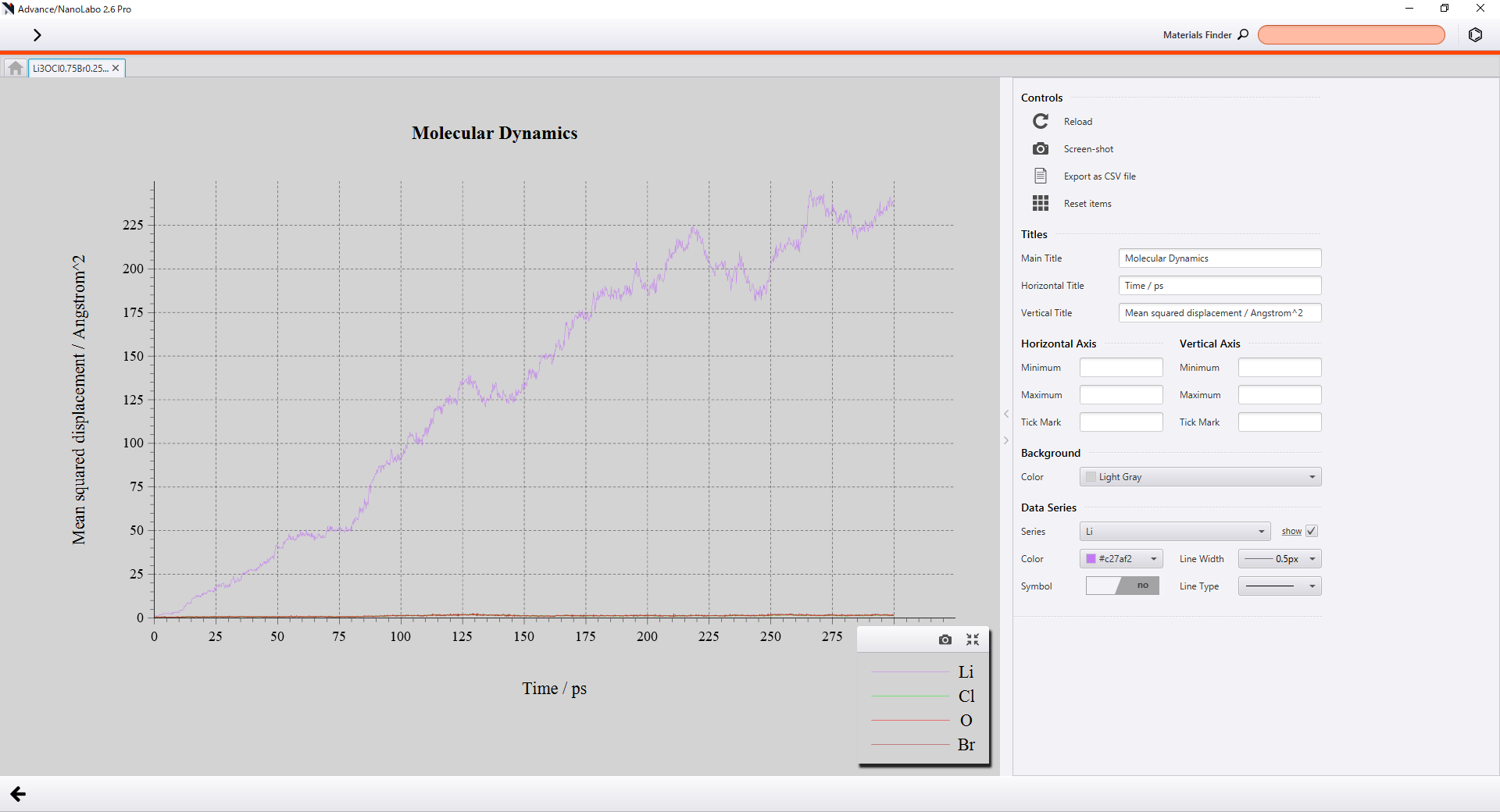
On Advance/NanoLabo, you can calculate and visualize MSDs of each element just by turning on Diffusion Coefficient button.
|
Visualization of MSDs on GUI |
Analysis results#
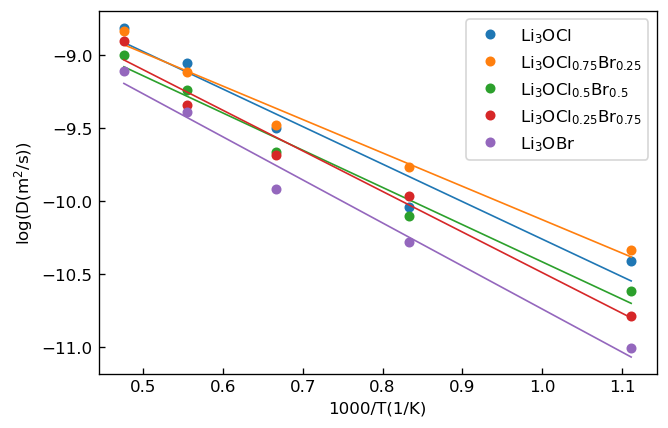
In a three-dimensional system, diffusion coefficient and MSD are related as
where is simulation time. We calculated diffusion coefficient of for each temperature by fitting the relationship to time dependence of MSD. We show Arrhenius plot of diffusion coefficient of obtained for each system below.
|
Arrhenius plot of diffusion coefficient D |
Ionic conductivity is calculated from diffusion coefficient and temperature as
where is number density of ion, is elementary charge, is Boltzmann constant and is activation energy.
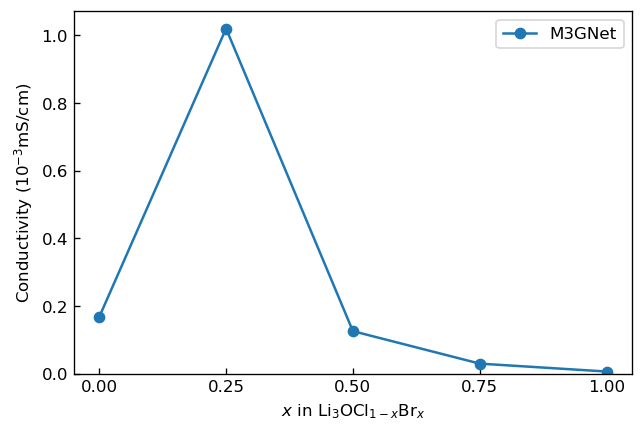
We calculated activation energy and the constant for each composition by fitting the latter relationship to the Arrhenius plot. Then, we calculated composition dependence of ionic conductivity from the obtained and . We show the result below.
We can see from the result that ionic conductivity increases to maximum value with increasing from 0 to 0.25, and monotonically decreases to minimum value with decreasing from 0.25 to 1 similarly to the result of ab initio molecular dynamics reported by previous study5. However, we note that the ionic conductivities of were underestimated compared to the result of ab initio molecular dynamics. The underestimation might have been caused by instability of M3GNet at high temperature leading overestimation of diffusion coefficient.
|
Composition dependence of ionic conductivity of Li3OCl1-xBrx |
Calculation of diffusion coefficient of a system at a certain temperature was done for about 2 hours without parallelization. It shows that molecular dynamics simulation with M3GNet has significantly low computational cost compared to ab initio molecular dynamics. Nevertheless, molecular dynamics simulation with M3GNet reproduced the tendency of composition dependence of ionic conductivity calculated via ab initio molecular dynamics.
関連ページ#
- ナノ材料解析統合GUI Advance/NanoLabo
- 解析分野:ナノ・バイオ
- 産業分野:材料・化学
- Advance/NanoLabo Product Information
- Advance/NanoLabo Documentation
-
We used LAMMPS (Large-scale Atomic/Molecular Massively Parallel Simulator) as a solver of molecular dynamics simulation. ↩
-
Chen, C., Ong, S.P. A universal graph deep learning interatomic potential for the periodic table. Nat Comput Sci 2, 718–728 (2022). ↩
-
Materials Project is an online database of substance materials. Materials Project is managed by Associate Professor Kristin Persson's group at University of California, Berkeley. ↩
-
Result of 2×2×2 super cell model with single vacancy () ↩
-
Deng, Z., Radhakrishnan, B., and Ong, S. P., Chemistry of Materials 2015 27 (10), 3749-3755 ↩↩↩
-
Chen, Ronghan, et al., ACS Applied Energy Materials 2021 4 (3), 2107-2114 ↩